J Pharm Pharmaceut Sci (www.cspscanada.org) 9(3):292-306, 2006
Drug Disease Interactions: Role of Inflammatory Mediators in Depression and Variability in Antidepressant Drug Response
Kenneth M. Kulmatycki and Fakhreddin Jamali
Drug Metabolism and Pharmacokinetics, Schering-Plough Research Institute, Kenilworth, New Jersey, USA and Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, Canada
Received, September 5, 2006; Revised, October 4, 2006; Accepted, October 5, 2006; Published,, October 5, 2006
Corresponding Author: Dr. K.M. Kulmatycki, Drug Metabolism and Pharmacokinetics, Schering-Plough Research Institute, Kenilworth, New Jersey, USA; kenneth.kulmatycki@spcorp.com , or Dr. F. Jamali, Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, Canada T6G 2N8, fjamali@ualberta.ca
ABSTRACT -- Increased cytokine expression and concurrent psychiatric symptoms were initially observed after administration of cytokines to patients afflicted with cancer, hepatitis and multiple sclerosis. Cytokines are a diverse group of soluble messenger proteins involved in the regulation, repair of cells, and control of immune events. During an inflammatory event expression of CD4+ T-lymphocyte helper (Th)1 cells that primarily produce pro-inflammatory cytokines is favored which can lead to development of inflammatory disease (e.g., cardiovascular disease). Similarly, relationships have been shown to exist between changes in inflammatory mediator concentrations, specifically pro-inflammatory cytokines, and depression. An increased prevalence of depression in patients afflicted with co-morbid inflammatory disease indirectly supports this association. In further support, antidepressants have been suggested to alleviate symptoms of depression via anti-inflammatory actions. Administration of anti-cytokines to patients with concurrent depression and inflammatory disease has resulted in relief of depressive symptoms. The exact role of inflammation in development of depression, however, remains to be determined. Nevertheless, increased expression of inflammatory mediators in depressed patients occurs which may lead to variability in response to antidepressant drug therapy. For example, depressed patients non-responsive to drug treatment are reported to have increased cell mediated immunity shown by elevated CD4+ T-cell activity, pro-inflammatory cytokine expression, and stimulation of the acute phase response. This suggests a psycho-neuroimmunological approach may be required for optimal pharmacotherapy.
Introduction
Conditions such as infection or injury stimulate an inflammatory response which is a complex cascade of non-specific immune events. Increased capillary blood flow and permeability that occurs during inflammation allows various cells of the immune system to enter the affected region resulting in swelling, redness, heat, and pain (1-3). This usually is a well controlled process resulting in removal of bacteria and tissue repair. However, excessive generation of mediators of inflammation such as cytokines by cells of the adaptive (T-lymphocytes) and innate (macrophages, monocytes, neutrophils) immune systems can lead to morbidity (2, 3). Cytokines are messenger proteins that are involved in cell regulation and repair as well as immunological responses to various stimuli (2-3). During an inflammatory event the balance between CD4+ T-lymphocyte helper 1 (Th1) cells that primarily produce pro-inflammatory cytokines [e.g. interferon (IFN)-g, interleukin (IL)-2, tissue necrosis factor (TNF-a)] and helper 2 (Th2) cells that mainly produce anti-inflammatory cytokines (e.g., IL-4, IL-10, IL-13) is altered favoring expression of Th1 cytokines (2-4). Secretion of pro-inflammatory cytokines by Th1 cells and cells of the innate immune system (e.g., macrophages) can result in development of disease (2-4). Increased inflammatory mediator expression (e.g., pro-inflammatory cytokines, C-reactive protein, nitric oxide) in association with morbidity is reported for various conditions such as cardiovascular, neurological and rheumatic disorders (5-9). Similarly, relationships have been shown to exist between changes in inflammatory mediator concentrations, specifically pro-inflammatory cytokines, and depression (10).
Materials and METHODS
INFLAMMATORY MEDIATORS AND DEPRESSION
Cytokines are large hydrophilic molecules thus penetration through the blood brain barrier is thought to be negligible and their role in pathology of psychiatric disorders limited. Cytokines may enter the brain, however, through regions where the blood brain barrier is absent (e.g., circumventricular organs), less restrictive (e.g. median eminence), compromised (e.g., multiple sclerosis) or by transport systems (10, 11). Inflammatory cytokines may also be produced within the CNS (e.g., IL-1, IFN-g, TNF-a) in the hypothalamus, hippocampus, basal ganglia and brain stem nuclei and bind to receptors on the surface of cells within the brain such as microglia, astrocytes and neurons (10, 11). In addition, by stimulating production of other mediators of inflammation such as nitric oxide and prostaglandins cytokines may indirectly influence CNS function (10-13).
Increased cytokine concentrations and concurrent psychiatric symptoms were initially observed after administration of cytokines to patients afflicted with cancer, hepatitis and multiple sclerosis (14-19). For example, to investigate the acute effects of cytokine therapy on development of depressive symptoms thirty-three cancer patients were separated into four treatment groups all receiving cytokine treatment. Each group was administered either subcutaneous IL-2 alone or in combination with IFN-a as low subcutaneous or high intravenous doses of IFN-a (20). Assessment of depressive symptoms and serum cytokine concentrations in these patients was conducted prior to beginning therapy and five days after starting treatment. The intensity of depressive symptoms was assessed using the Montgomery and Asberg Depression Rating Scale (MADRS). On these same days blood samples were collected from each patient for measurement of serum cytokine concentrations (IL-6, -10, -1ra) and receptors [sIL-2r and (leukemia inhibitory factor-receptor) LIF-R]. No differences in MADRS scores and cytokine levels were found between the treatment groups prior to starting immunotherapy. IL-2 treatment alone and not INF-a treatment alone significantly raised the depression scores. Combined treatment of IL-2 and IFN-a resulted in higher depression scores than IL-2 alone as shown in Figure 1. Compared to patients that received IFN-a alone, patients treated with IFN-a and IL-2 had a greater stimulation of the cytokine network. In the IFN-a treated patients increases in IL-1ra and LIF-R were more pronounced after intravenous administration. Increased serum levels of IL-10 in patients that have a significant increase in MADRS scores indicate the presence of an inflammatory response. Increases in cytokine concentrations were associated with development of other toxic effects such as fever and nausea/vomiting that were attributed to pyrogenic and digestive/anorexia effects of cytokines. In addition, age, gender, medical/pharmacological factors did not contribute to the variation in IFN-a and IL-2 expression in patients or MADRS scores during cytokine treatment.
The observed elevation of pro- and anti-inflammatory cytokines in the above cancer patients (20) after administration of IL-2 alone, IL-2 and IFN-a, and high dose intravenous IFN-a therapy demonstrates immune activation. This suggests an association between development of depressive symptoms and activation of the immune system (i.e., inflammation) in cancer patients undergoing cytokine therapy. In further support, the development of acute anxiety and depression was evaluated in cancer patients treated with subcutaneous IL-2 (n=20), IFN-a2b subcutaneous (n=8) or intravenous (n=14) or a combination of subcutaneous IL-2 and IFN-a2b (n=6) using the Covi Anxiety Scale and MADRS scores prior to treatment and on day 3 and 5 of cytokine therapy. In further support, the development of acute anxiety and depression was evaluated in cancer patients treated with subcutaneous IL-2 (n=20), IFN-a2b subcutaneous (n=8) or intravenous (n=14) or a combination of subcutaneous IL-2 and IFN-a2b (n=6) using the Covi Anxiety Scale and MADRS scores prior to treatment and on day 3 and 5 of cytokine therapy. Depression scores were significantly increased after IL-2 and not IFN-a2b treatment on day 5 (21). Patients treated with IL-2 and IFN-a2b had higher depression scores by day 5 compared to those treated with IL-2 alone. Anxiety scores were significantly increased only for patients treated with IL-2 combined with IFN-a2b which may have been due to somatic effects of these two cytokines. However, MADRS scores for these patients were positively correlated with Covi scale scores indicating that depressed mood overlapped with anxiety in these patients. Development of depressive and anxiety symptoms were suggested to be due to IFN-a2b acting synergistically with IL-2 treatment amplifying the neuropsychiatric effects of IL-2.
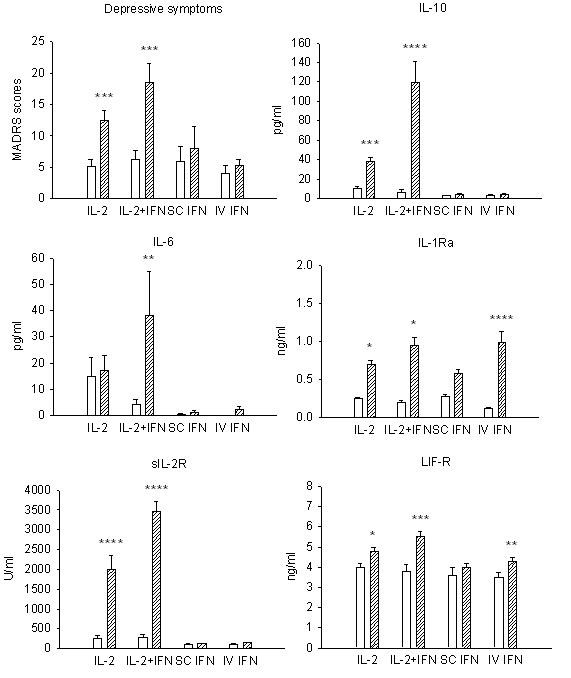
Figure 1. Patients treated with IL-2 or IL-2 plus IFN-a have increased cytokine levels and concomitant mood disturbances during treatment. Increased IL-10 expression reflects presence of an inflammatory response. Changes in MADRS scores and cytokine serum concentrations between baseline (blank bar) and fifth day of therapy (stripped bar) in each treatment group [IL-2 (n=13), IL-2 + IFN (n=5), SC IFN (n=5), IV IFN (n=10)] means ± SEM. ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05. From reference 20 with permission.
Development of depressive illness after administration of pro-inflammatory cytokines may be due to the various roles that cytokines have in facilitation of communication between the immune and central nervous systems (22-28). For example, IL-1b, IL-6 and TNF-a are key mediators in immune-to-brain communication by orchestrating responses such as fever and changes in sleep patterns (29-31). In addition, during the course of an infection physiological and psychological effects of immune activation occur that are primarily mediated by the central actions of peripherally released inflammatory cytokines (e.g., IL-1a, -1b, IL-6, TNF-a) referred to as sickness behavior (32). Sickness behavior is accompanied by fever and behavioral changes such as decreased appetite, anorexia, weight loss, fatigue, sleep disturbances, impaired cognitive abilities, dysphoria, anhedonia and depressed mood which are all symptoms of depression (11, 32). This behavior appears to be a reorganization of the priorities of the patient to cope with infectious pathogens and inflammation. Individuals also experience these same symptoms after systemic or central administration of cytokines (14-18, 33, 34). Administration of endotoxin to humans, interestingly, at doses below those known to cause illness increased IL-6 and TNF-a expression resulting in transient anxiety, depression, and memory impairment. The symptoms of mental illness were positively correlated with changes in expression of IL-6 and TNF-a (35).
Patients administered cytokines generally experience flu-like symptoms such as fever, malaise, headache, myalgia at the beginning of immunotherapy that usually diminishes as treatment is continued. Psychiatric disorders (e.g., dysphoria, anhedonia, anxiety and impaired cognitive ability) generally occur later suggesting symptoms of psychiatric illness are not secondary to physical discomfort. Administration of cytokines has been linked to various psychiatric conditions ranging from subtle memory and attention impairments to delirium, psychosis and suicide (16-18, 34, 36). Changes in mood and cognitive disturbances in some individuals may even persist for weeks to months after finishing or stopping treatment (36). Determining the role of immune system activation in pathology of depression whether idiopathic or due to pro-inflammatory cytokine administration, however, is difficult due to heterogeneous patient sampling when comparing studies. Nevertheless, immune changes in association with depressive symptoms are reported such as increased pro-inflammatory cytokines and receptors (e.g., sIL-1ra, sIL-2r, IL-6, sIL-6r), prostaglandin E2, and thromboxane B2 (37-43). In addition, activation of the acute phase response, a systemic response to local inflammation, shown by increased concentrations of a1-acid glycoproteins, antitrypsin, antichymotrypsin, hatoglobin, C-reactive protein, celuroplasmin and decreased concentrations of albumin is reported to occur in depressed individuals (37, 43-49).
Depression is estimated to afflict 121 million people worldwide (50). Depression and anxiety are often concurrent conditions especially in situations where exposure to psychological stress is chronic and/or where physiological responses to stressors are disproportionately large (51, 52). This raises the question of whether anxiety may contribute to the inflammatory mediator over-expression that is observed in depressed individuals.
To investigate whether psychological stress alters pro-inflammatory cytokine production concentration of various cytokines were measured from blood samples obtained from medical students one month before and after as well as one day before an academic examination (53). Students were divided into two groups those who had low stress perception and those who perceived themselves to be highly stressed and anxious. Blood samples for cytokines were taken before (mean of 45 ± 6.0 days before), after (mean of 36 ± 5.9 days after) and stress (one day before) a difficult academic examination. Psychological stress was associated with increased concentrations of TNF-a, IL-6, IL-1ra, IFN-g and IL-10. The students who had high stress perception had significantly greater concentrations of TNF-a, IL-6, IL-1ra and IFN-g than those with low stress perception as shown in Figure 2. Students with high anxiety response had higher pro-inflammatory IFN-g and lower anti-inflammatory IL-10 and IL-4 concentrations compared to students with lower anxiety. Increased concentrations of pro-inflammatory cytokine expression accompanied perception of psychological stress indicate stress-induced anxiety may be associated with an increase in Th1 cell responses.
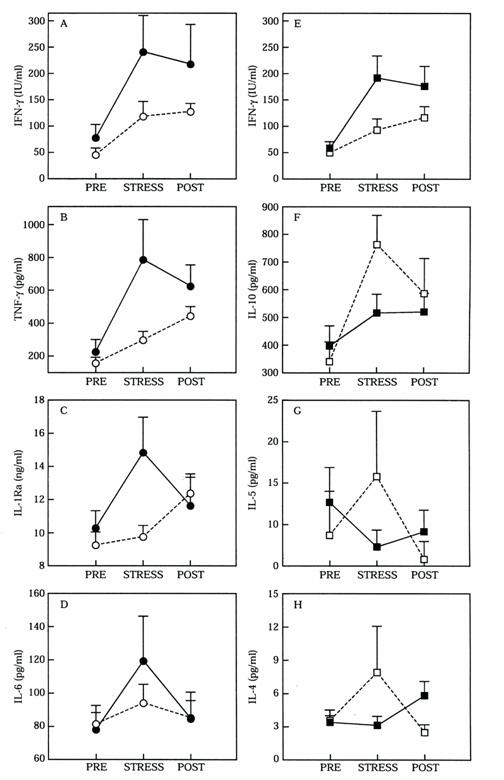
Figure 2. Measurements of serum cytokines greater than one month before (PRE) and after (POST), and one day before (STRESS) a difficult academic examination. The medical students were divided depending on their stress perception as high (n) and low (O) (A-D) and students with high (g) versus low (c) anxiety responses (E-H). Values are mean and standard error of the mean. From reference 53 with permission.
Similar cytokine patterns are reported for individuals afflicted with anxiety or depression. For example, mean plasma concentrations of IL-18, a pro-inflammatory cytokine, was three-fold higher in non-medicated patients with major depression and four fold higher for patients with panic disorder compared to normal controls (54). Although speculative, it is probable that psychological stress by contributing to increased inflammatory mediator expression may influence onset and progression of depression (55).
MECHANISM OF INVOLVEMENT OF INFLAMMATORY MEDIATORS IN DEPRESSION
Disturbances in neurochemical transmission and modulation of the hypothalamus-pituitary-adrenal (HPA) axis are mechanisms proposed by which cytokines may be involved in pathogenesis of depression. Depressive illness is typically characterized as a disorder of noradrenaline and serotonin neurotransmission with treatment focused on normalizing neurotransmitter function (56, 57). Serotonin is synthesized from tryptophan, an essential amino acid produced peripherally, that moves from the systemic circulation into the brain via active transport and then converted to serotonin. Various mechanisms have been suggested by which pro-inflammatory cytokines may alter neurotransmitter function. To date these include, reduction in tryptophan availability for serotonin synthesis by activating tryptophan degrading enzyme indoleamine-2,3-dioxygenase and stimulation of amino acids that compete with tryptophan for uptake into the CNS (10, 11, 58-60). Cytokine-induced stimulation of indoleamine-2,3-dioxygenase, an enzyme that switches the synthesis of serotonin from tryptophan to kynurenine and quinolinic acid, results in a reduction of serotonin synthesis (61, 62). It is probable that decreased production of serotonin by this mechanism may lead to development of depression. For example, cancer patients treated with cytokine therapy had decreased serum tryptophan levels during the course of therapy with the degree of tryptophan reduction correlating with severity of depressive symptoms (63). In addition, a significant increase in the kynurenine/tryptophan ratio was associated with inflammatory signs in patients afflicted with hepatitis C undergoing IFN-a therapy. These patients’ also experienced significant increases in MADRS and Hamilton Anxiety scores (64). Quinolinic acid produced from kynurenine is a neurotoxic substance that has been implicated in neurodegenerative disorders. Increased activity of the kynurenine pathway stimulated by pro-inflammatory cytokines is reported to result in production of neurotoxic metabolites in depressed individuals (59, 65-68).
Cytokines have also been proposed to induce depression by modulation of the HPA axis which is a major mechanism by which the brain reacts to stress. When subjected to stress neurons in the paraventricular nucleus of the hypothalamus secrete corticotropin-releasing-factor which in turn stimulates the release of adrenocorticotropin (ACTH) from the anterior pituitary. ACTH then stimulates the synthesis and release of glucocorticoids (e.g., cortisol) from the adrenal cortex. Glucocorticoids by regulating paraventricular nucleus and hippocampal neurons have a powerful negative feedback on the HPA axis and under normal circumstances increased levels enhance hippocampal inhibition of HPA activity. In pathological states, however, sustained elevated levels of glucocorticoids may damage hippocampal neurons reducing the inhibitory control that the hippocampus exerts on the HPA axis resulting in further elevation of circulating glucocorticoid levels and subsequent hippocampal damage (69). Excessive HPA axis activity is observed in approximately 50% of individuals afflicted with depression which is usually corrected by antidepressant treatment (69). Increased levels of cortisol in depressed patients may be toxic to hippocampal neurons contributing to cognitive abnormalities of depression (69-71). IL-1 is reported to impair glucorcorticoid receptor translocation and function suggesting that cytokines may also mediate glucorcorticoid resistance by direct effects on the glucocorticoid receptor (72). Pro-inflammatory mediator alterations in neurotransmitter function may also influence HPA axis function. Cytokine-induced activation of indoleamine-2,3-dioxygenase may impair the negative feedback of corticosteroids on the HPA axis through production of quinolinic acid, a potent N-methyl-D-aspartate receptor agonist, causing hippocampal atrophy and loss of glucocoricoid receptors (73). Psychological stress similar to depression is reported to be accompanied by HPA axis hyperactivity and increased catecholaminergic turnover. Increased levels of pro-inflammatory cytokines induced by stress may contribute to the glucocorticoid resistance and HPA axis hyperactivity in depressed individuals (27, 65, 74-75). Pro-inflammatory cytokine effects on neurotransmitter function and the HPA axis are probable explanations for involvement of inflammation in development of depression.
Several theories have been proposed to explain the relationship between inflammation and development of depression, for example, “the inflammatory response system model of major depression”, “macrophage theory of depression”, and “the cytokine hypothesis of depression” (76-78). In these models depression is considered to be a psychoneuroimmunological disorder due to peripheral immune activation through release of pro-inflammatory cytokines that are proposed to be responsible for a variety of behavioral, neuroendocrine and neurochemical alterations in depressed patients. Despite numerous reports of associations between depressive illness and pro-inflammatory cytokine over-expression, it remains to be determined whether pro-inflammatory cytokines have a causal role in the pathogenesis of depressive illness or just sustain symptoms, as it is unclear whether cytokine abnormalities precede or follow the onset of illness (11, 79). This is likely due to the fact that most studies investigating the relationship between immune activation and depression are correlational using animal models or in vitro assays. In human studies, heterogeneous patient populations, differences in disease severity, small sample sizes and treatment with and without antidepressants may also be factors leading to discrepancies between studies. In addition, many investigations assessing the relationship between pro-inflammatory cytokine expression and depressive disorders involve administering relatively high doses of cytokines either centrally or systemically. This raises the concern of whether lower concentrations of cytokines would influence brain function to the same extent. It is likely that lower concentrations affect the CNS to a similar degree as higher concentrations at least on sleep-wake behavior (80).
In contrast, considerable evidence exists indicating that cytokines play a role in the pathogenesis of depression. Cytokines are available as well as produced in the CNS and influence cerebral functions through binding to receptors on various tissues of the brain. Immune therapies are frequently accompanied by psychiatric side effects that usually remit only after either discontinuation therapy or antidepressant treatment. Antidepressant drugs are reported to attenuate the behavioral and neuroendocrine effects of immune activation in depressed patients. Depression is frequently a co-morbid disorder with various inflammatory disorders (e.g., cancer, rheumatoid arthritis, cardiovascular disease). In addition, administration of anti-cytokines (e.g., anti-TNF-a) to patients afflicted with depression and chronic inflammatory conditions (e.g., Crohn’s disease) resulted in alleviation of depressive symptoms not related to changes in physical discomfort and lifestyle. Irregardless of the role of cytokines and other mediators of inflammation in depressive disorders increased expression of mediators of inflammation in depressed patients occurs. It is probable that over expression of mediators of inflammation similar to other inflammatory disorders may influence pharmacotherapy (9).
DEPRESSION IN PATIENTS WITH OTHER INFLAMMATORY CONDITIONS
A higher than typical prevalence of depression occurs in patients afflicted with inflammatory disorders (e.g., rheumatoid arthritis) indirectly supporting an association between increased pro-inflammatory mediator expression and pathophysiology of depression (81). Hence, depression in rheumatic individuals may not merely be a psychological response to pain and incapacitation due to immune activation (81). Similar to arthritic patients, depression is more prevalent than in the general population for individuals afflicted with cardiovascular disorders especially with regard to heart failure patients (82). In a recent study, serum concentrations of pro-inflammatory (TNF-a, IL-6) and anti-inflammatory (IL-10) cytokines and soluble apoptosis mediators (Fas/Fas ligand) were compared between depressed and non-depressed patients with mild to moderate stable chronic heart failure. Depressive symptoms were assessed using the Zung Self-Rating Depression Scale (83). Patients with depression exhibited significantly higher levels of TNF-a, soluble apoptosis mediator Fas ligand (sFasL), and lower levels of anti-inflammatory cytokine IL-10 compared to patients without depressive symptoms as shown in Figure 3.
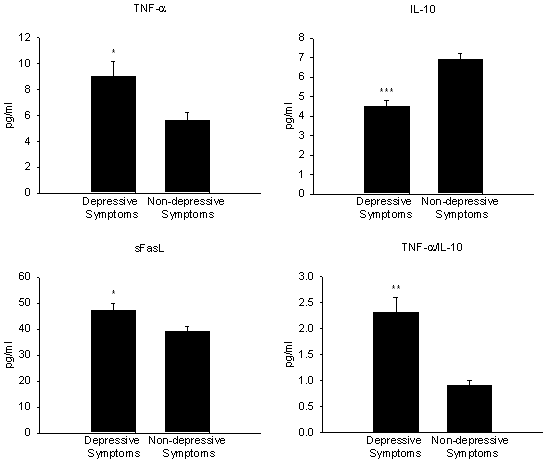
Figure 3. Differences in plasma levels of circulating TNF-a, IL-10, sFasL and TNF-a/IL-10 ratio in depressed (n=15) and non-depressed (n=20) patients afflicted with mild to moderate stable chronic heart failure. Values are represented as means ± SEM. ***p<0.001; **p<0.01; *p<0.05. Adapted from reference 83 with permission.
The TNF-a/IL-10 ratio in depressed patients was significantly correlated with the severity of depressive symptoms. No differences in IL-6 serum concentrations were observed between the two groups which may be due to IL-6 having a limited role in neurohormonal reactions associated with depression in mild to moderate heart failure patients. The authors rationalized these findings by an altered balance between pro- and anti-inflammatory cytokines resulting in abnormal central metabolism of serotonin and enhancement of catecholaminergic and other critical pathways in specific regions of the brain associated with depression such as the paraventricular nucleus of the hypothalamus. In addition, in these patients central neurohormonal abnormalities may be essential for development of emotional distress that may further enhance abnormal peripheral immune responses by inducing release of pro-inflammatory factors associated with HPA axis hyperactivity. Increased inflammatory response in heart failure patients with concurrent depression may potentially impact patient outcome. Depression in heart failure patients has in fact been independently associated with poor prognosis (82, 84). Depression and co-morbid disease is of particular relevance to the elderly as pro-inflammatory cytokine expression and incidence of multiple conditions is report to increase with aging (85, 86). For example, a preliminary report indicates higher levels of pro-inflammatory cytokine (e.g., IL-1β) correlate with severity of depressive symptoms in older patients (87).
TREATMENT OF DEPRESSION CAUSED BY CYTOKINES
Pharmacological treatment of depression secondary to cytokine administration has included antidepressants, opiate antagonists, stimulants and corticosteroids (15, 88-94). Serotonin selective reuptake inhibitors as well as tricyclic antidepressants have been used to treat and prevent IFN-a-induced depression (95, 96). For example, forty patients afflicted with malignant melanoma were treated with IFN-a, patients received placebo (n=20) or paroxetine (n=20) from two weeks prior to initiation of interferon therapy up to twelve weeks of interferon treatment (97). Comparing the two groups, 35% of the placebo group and only 5% of the paroxetine group had to discontinue IFN-a therapy due to depression. The incidence of other side effects was similar in the two groups. Antidepressant therapy had a significant effect on severity of symptoms for anxiety and depression that were lower in paroxetine treated group than placebo. Paroxetine treatment resulted in patients free from depression and decreased the likelihood for discontinuation of IFN-a therapy due to severe depression as shown in Figure 4. Screening for symptoms of anxiety and depression prior to initiating IFN-a treatment was suggested to aid in identifying patients at high risk for development of psychiatric complications during immunotherapy which would reduce the number of patients discontinuing treatment.
Administration of anti-depressant drugs is reported to result in immune system changes (i.e., decreasing expression of inflammatory mediators) suggesting a link between increased inflammatory mediator expression and disease (98). In support, cytokine antagonists have been administered to patients with chronic inflammatory disorders to alleviate physical illness as well as symptoms of concurrent depression. Administration of anti-cytokines (e.g., TNF-a) for treatment of patients afflicted with Crohn’s disease and concurrent depression resulted in alleviation of physical and depressive symptoms. Anti-TNF-a treatment significantly improved the quality of life in these patients resulting in increased ability to work and participate in social activities, decreased fatigue and depression (99). Similarly, treatment with anti-TNF-a is reported to clear up psoriatic lesions and relieve symptoms of fatigue and depression in patients with psoriasis (100). Although speculative, this suggests an association between increased pro-inflammatory cytokines and pathogenesis of depressive disorder.
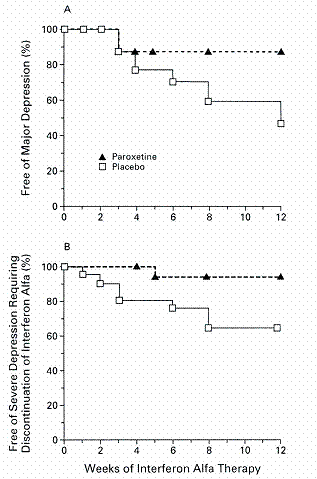
Figure 4. Kaplan-Meier analysis of the percentage of patients in the placebo and paroxetine groups who were (A) free of major depression and (B) of severe depression requiring the discontinuation of IFN-a treatment. Paroxetine significantly decreased the incidence of major depression in patients receiving IFN-a therapy. From reference 97 with permission.
INFLAMMATION AND VARIABILITY IN DRUG RESPONSE
Clinical improvement after treatment with antidepressants generally occurs in less than 70% of depressed patients with relapses as high as 50% (101, 102). Ironically there is a paucity of information regarding the etiology of treatment resistant depression despite such a high occurrence. Activation of cell-mediated immunity may be one probable explanation for decreased response to drug therapy. For example, patients non-responsive to antidepressants had significantly higher percentage of CD4+ T-cells and CD4+/CD8+ T cell ratio and significantly lower percentage of CD8+ T cells than patients responsive to therapy and normal controls (103). Treatment resistant patients are reported to have elevated plasma levels of IL-6, sIL-6r, IL-1ra and sIL-2r (37, 40). In addition, B cell dependent antibody production directed against serotonin and gangliosides was significantly higher in patients resistant to treatment compared to healthy volunteers and responders to therapy (37). This suggests a chronic activation of the inflammatory response system that is more pronounced in patients not responding to pharmacotherapy. Illness severity and length, increased stress perception, maladaptive coping strategies, reduced eating and sleeping and weight loss may contribute to the immune alterations in depressed patients resistant to treatment. In fact, increased cytokine production in depressed patients has been correlated with chronicity of illness (104).
Acute phase proteins are produced during the acute phase response, a systemic response to inflammation, such as a1-acid glycoprotein and reported to be increased in patients that have a longer duration of depressive illness and refractory to therapy (3, 37, 49). Drug therapy has been shown to influence acute phase protein expression. For example, the effect of lithium on plasma levels of acute phase proteins and antidepressant effectiveness was evaluated in patients with refractory depression (105). Thirty-two patients with refractory depression defined as failure to respond to at least two antidepressant treatments with the duration of each treatment six weeks were treated with an antidepressant plus lithium. Plasma concentrations of acute phase proteins a-1 acid glycoprotein (APG), a-1 antichymotrypsin (ACT), C-reactive protein, and microheterogenity products of APG and ACT were determined (AGP-RC and ACT-RC). Microheterogenity products are plasma proteins in which modifications in glycosylation occurs in the liver mediated by pro-inflammatory cytokines such as IL-1, -6, TNF-a, interferon, and glucocorticoids. Before lithium treatment, non-responders had increased levels of C-reactive protein, AGP-RC and ACT-RC compared to responders. After lithium therapy in responders a significant decrease in all acute phase proteins was observed while non-responders continued to have higher AGP-RC concentrations and less of a decrease in a-1 acid glycoprotein plasma concentrations compared to those that responded to treatment. The authors state that higher values of AGP-RC observed for non-responders has also been reported for patients with burns, rheumatoid arthritis and lupus erythematosus with infection and changes in the glucocorticoid status was speculated for non-responders. In support, lithium has been shown to have immunoregulatory effects and suggested to normalize disequilibrium between pro- and anti-inflammatory modulators (106). Although further studies need to be conducted, it is probable that depressive illness resistant to treatment may be accompanied by chronic immune activation that is more pronounced in patients that do not respond to drug therapy.
CONCLUSION
The association between increased expression of pro-inflammatory cytokines and pathology of depression is, at most, controversial and remains to be unequivocally understood. Nevertheless, whether cause or consequence, increased expression of pro-inflammatory mediators in depressive disorders occurs and may influence pharmacotherapy. This may explain treatment resistance in some patients, however, at the same time complicates the dose-response relationship as additional considerations such as the immune status of the individual may need to be considered. It is possible that involvement of pro-inflammatory cytokines in the pathology of depression is complex, for example, many cells have receptors for the same cytokines and cytokines tend to have overlapping functions leading to formation of networks with cascade responses. The effect of pro-inflammatory cytokine over-expression in pathogenesis of depressive disorders, therefore, may be due to combined activities of many cytokines or perhaps in combination with other mediators of inflammation (107). Associations between decreased response to drug and increased expression of pro-inflammatory mediators (i.e., inflammatory status) are reported for many other diseases (9). Decreased response to antidepressant medication may be especially relevant to the elderly who are reported to have increased expression of inflammatory mediators and incidence of multiple conditions. A psychoneuroimmunological perspective on the treatment of depressive illness may be required for successful treatment and is suggested to change current views on depression (108). Understanding the role that pro-inflammatory mediators play in response to drug therapy in depressed patients, therefore, may aid in explaining treatment resistance and assist in designing more effective pharmacotherapy.