J Pharm Pharmaceut Sci (www.cspscanada.org) 9(3):307-316, 2006
Evaluation of in vitro-in vivo correlation and anticonvulsive effect of carbamazepine after cogrinding with microcrystalline cellulose
Mohammad Barzegar-Jalali1, Alireza Mohajjel Nayebi, Hadi Valizadeh, Jalal Hanaee, Azim Barzegar-Jalali, Khosro Adibkia, Mahdieh Anoush, Mohammad Sistanizad
Department of Pharmaceutics, Faculty of Pharmacy and Drug Applied Research Center, Tabriz University of Medical Sciences, Tabriz, Iran. Department of Pharmacology and Toxicology, Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran. Department of Medicinal Chemistry, Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran. Iranian Blood Transfusion Organization, Ardabil, Iran Pharmaceutical Nanotechnology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran. Department of Clinical Pharmacy, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran.
Received, May 31, 2006, revised, September 6, 2006; accepted October 4, 2006; published October 19, 2006.
Corresponding Author: Dr. Mohammad Barzegar-Jalali, Department of Pharmaceutics, Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran. E-mail: barzegar_jalali@yahoo.com
ABSTRACT -- Purpose. Carbamazepine is a poor water soluble drug and its bioavailability is limited by dissolution rate. Dissolution, serum concentration and anticonvulsive effect of the drug have been evaluated after cogrinding with microcrystalline cellulose. A cogrinding technique was used to increase the dissolution, serum concentrations and anticonvulsive effect of the drug. A novel deconvolution technique of in vitro- in vivo correlation was evaluated. Methods. The drug coground with microcrystalline cellulose, the corresponding physical mixture, unground and ground drug powder were subjected to dissolution measurement. Coground and unground drug serum concentrations were investigated in rabbits. Also the anticonvulsive effects of the latter preparations were assessed in mice. For elucidation of observed in vitro and in vivo differences FT-IR spectroscopy, X-ray diffraction patterns and DSC thermograms of the preparations were studied. Results. The dissolution of the coground was the highest (percent dissolved in the first 20 minutes, %D20’, was 97.5). The unground drug powder exhibited the lowest dissolution (%D20’=40). The difference was reflected in their corresponding area under the mean serum concentration curves between 0-16 hr (118.96 vs 54.17 μg.hr/ml) as well as protection abilities against strychnine and electrically induced seizures. The onset of tonic seizures induced by strychnine was increased between 40-140 % in the case of the coground system depending on dose and time of carbamazepine administration. Conclusion. Cogrinding was an effective technique in increasing carbamazepine dissolution due to reduced crystallinity as seen in X-ray pattern, enhanced wettability and decreased particle size, which in turn resulted in increased serum concentrations and its anticonvulsive effect. A novel simple deconvolusion technique not requiring intravenous data denoted as the double reciprocal area method was used to establish correlation between in vitro and in vivo parameters.
Introduction
Carbamazepine is a dibenzazepine derivative with antiepileptic and psychotropic properties. It is also used in the treatment of trigeminal neuralgia and pain associated with other neurological disorders (1). It belongs to class II biopharmaceutical classification system (BCS) which is characterized by high membrane permeability, slow dissolution rate due to low aqueous solubility, and high peroral dose (2). Therefore, bioavailability rate of this drug is limited by the dissolution rate. Several attempts have been made to increase the dissolution or the bioavailability of the drug (3-12). Among the various techniques of enhancing dissolution of poorly water soluble drugs the cogrinding method has been exploited for drugs such as naproxen (13-15), celecoxib (16), triamterene (17) iboproxam (18) and dehydroepiandrosterone (19). However literature survey showed that the methodology has not been utilized for the augmentation of carbamazepine dissolution rate and bioavailability. The cogrinding is economically and environmentally desirable as unlike other techniques it does not require toxic solvents (6) and sophisticated equipments (9). In present work we applied the cogrinding method to increase carbamazepine dissolution rate using microcrystalline cellulose as the hydrophilic carrier and the physicochemical characteristics of prepared coground system was compared with those of the physical mixture as well as ground and unground drug powder using powder x-ray diffraction, Fourier transform infra red spectroscopy, and differential scanning calorimetery. Furthermore, the bioavailability in rabbits and anticonvulsive effect in mice of the coground system and unground drug were investigated and a novel level A (point by point) in vitro-in vivo correlation was established successfully.
Materials and METHODS
Carbamazepine (Arastou Co., Tehran, Iran), tolybarb, which is 5-ethyl-5-(p-methylphenyl) barbituric acid (Aldrich Chemical Co., Milwaukee, USA), potassium dihydrogen phosphate, disodium hydrogen phosphate, sodium hydroxide, HPLC grade methanol, chloroform, and isopropanol (Merck, Darmstadt, Germany), microcrystalline cellulose (Avicel RC591, FMC, Brussels, Belgium).
Preparation of coground system, physical mixture and ground drug
Coground system with 1:1 W/W (final weight 20 g) carbamazepine to microcrystalline cellulose was prepared using ball mill (Fritsch, Germany). The volume of the mill chamber was 300 ml. Balls of different diameter ranging from 8 to 20 mm occupying one third of volume of the chamber were employed. The vibration rate was set to 360 rpm. The grinding time was 3 hours. Similar methodology was utilized to prepare the ground carbamazepine powder.
A physical mixture containing one part drug and one part carrier was prepared using the bottle method. The drug contents of the coground system and physical mixture were determined spectrophotometricaly at 286 nm (UV-160, Shimadzu, Kyoto, Japan) after dissolving in 5 percent hydromethanolic solution and filtration. The measured contents were 99.3±0.5 percent of the expected value.
Dissolution studies
The dissolution of carbamazepine unground and ground powder as well as its physical mixture and the coground system was studied using USP apparatus No. 2. The amount of drug in the sample was 10 mg which was added to 900 ml of distilled water as dissolution medium. Under such circumstances a perfect sink condition was maintained to mimic the dynamic situation in GI tract. The mixture was stirred at 100 rpm at 37±0.3 ◦C. 5 ml samples were withdrawn at predetermined times, filtered and assayed at 286 nm on the spectrophotometer. The drug concentration in the samples was corrected considering the concentrations of the previous samples. Each dissolution test was carried out in triplicate.
X-ray crystallography, IR spectroscopy and Differential scanning calorimetery
X-ray diffraction patterns of the samples were obtained using an automatic powder diffractometer (Siemens- 850, Munich, Germany) using Cu Kα radiation at a scan rate of 2° min-1 in terms of 2θ angle. The KBr disk sample preparation technique was used to obtain the IR spectra of the formulations on an IR spectrophotometer (FTIR 3400 Shimadzu, Kyoto, Japan). The amounts of sample and KBr, the scanning range of wave number and the resolution were, 5 mg and 45mg, 400-4000 cm-1 and 1 cm-1, respectively. Differential scanning calorimetery (DSC) was performed on a Shimadzu DSC60 (Kyoto, Japan). The samples (5 mg) were analyzed in covered aluminum pans with a heating rate of 50°C min-1 in the range of 30-250 °C.
In vivo studies
The pharmacokinetic experiments were carried out in albino rabbits, 2.0 ± 0.5 kg (1-1.5 years age), housed in standard cages, one per cage. The permission for animal studies was obtained from the Ethics Committee of the University. Eleven rabbits were involved in the study to investigate kinetics of carbamazepine unground powder and carbamazepine-microcrystalline cellulose coground. Twelve hours prior to experiments the animals were fasted but had free access to water. All animals received orally a single dose of carbamazepine (equivalent to 120 mg/kg) as unground and coground suspended in 10 ml of distilled water on treatment days 1 and 2, respectively, with a one week wash out period between two successive dosings. Blood samples were collected from the marginal vein by individual venous puncture pre dose (0 hr) and at 1, 2, 3, 4, 5, 6, 8, 10, 12, and 16 hr post dose. The clotted samples were centrifuged at 5500 rpm for 8 min. The serum samples were stored at -20°C until analysis and a well established HPLC method (20) was used to determine carbamazepine serum concentration.
The drug was extracted from serum sample by adding 100 µl phosphate buffer (pH=6.8) and 1.5 ml of extractant (5% isopropanol in chloroform) containing tolybarb as an internal standard. After centrifugation at 5500 rpm for 5 min the organic phase was collected and evaporated using nitrogen gas. The dried residue was dissolved in 50 µl of mobile phase consisting phosphate buffer 1300 ml, methanol 350 ml and acetonitril 350 ml. 15 µl of resulting solution was injected onto the HPLC column. Chromatography was performed on a reverse phase phenomenex ODS column (5 µm, 250 ´ 4.6 mm I.D.) and guard column (ODS 20 ´ 4.6 mm). The flow rate was 2 ml/min. The effluent was monitored at 204 nm with a variable wavelength UV detector (Cecil, England). The latter wavelength differed from the one used in the dissolution studies which was due to difference in the nature of solvents utilized. Chromatograms were integrated and analyzed using a computerized data acquisition system (Cecil, England). The retention time for tolybarb and carbamazepine were 4.8 and 6.68 min respectively. A linear calibration plot was obtained using the drug to internal standard peak area ratio in the concentration range of 1-16 μg/ml. The plot was used to determine the drug serum concentration.
In vitro-in vivo correlation
The double reciprocal area method (25) has been advocated to establish a quantitative correlation between in vivo and in vitro parameters of the drug in the form of the following non-linear relationship
![]() (1)
(1)
Where ![]() ,
, ![]() , m, b and j are the areas under the drug serum concentrations and percent drug dissolved vs normalized time (tn) curves between 0 and tn and constants respectively. The normalized time is the ratio of any in vivo and in vitro sampling time with respect to the corresponding arbitrary last sampling time.
, m, b and j are the areas under the drug serum concentrations and percent drug dissolved vs normalized time (tn) curves between 0 and tn and constants respectively. The normalized time is the ratio of any in vivo and in vitro sampling time with respect to the corresponding arbitrary last sampling time.
The normalization of time is necessary to bring in vivo and in vitro times to the same scale. The percent dissolved corresponding to each in vivo value of tn was obtained via interpolation of the normalized dissolution curve.
Anticonvulsion study
For assessment of anticonvulsant activity, male NMRI mice (Pasteur Institute of Iran) weighing 24-26 g were used. Animals were kept under a 12-hr light-dark schedule at an ambient temperature of 23 ± 3°C, and allowed freely food and water. The work was authorized by the Ethics Committee of the University.
The antiseizure effect of the coground carbamazepine with microcrystalline cellulose was compared to that of unground carbamazepine, using strychnine (STR)-induced seizure and maximal electroshock (MES) as two commonly used seizure models. Twenty mice were randomly and equally divided into two groups. In order to perform STR-induced seizure test, STR at a dose of 1.7 mg/kg was injected intraperitonealy 3 and 6 hours after peroral doses of 2 and 2.5 mg of each formulation. The animals were monitored for 10 min post STR injection to determine the initial latency time, wild running, tonic seizures, respiratory arrest and mortality.
The MES test with suprathreshold stimulation was carried out via ear clip electrodes by means of a stimulator that delivered a fixed current of 100 mA with a pulse frequency of 128 Hz for 0.2 seconds. The electrodes were placed in 0.9% sodium chloride solution before application. The duration of tonic hind limb extension (limb extension exceeding a 90° angle with the plane of the body) was used as the criterion of convulsion.
Statistical analysis
Paired Student t-test, one-way ANOVA, Tukey test and regression analyses were performed according to usual procedures (21). Accuracy of predicted in vivo data was assessed by:
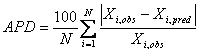 (2)
(2)
APD is average percent deviation of predicted value, Xi,pred, from its observed value, Xi,obs and N is the number of data. Xi is either the partial area under the curve or drug serum concentration.
RESULTS
Dissolution
The dissolution profiles of unground carbamazepine, its 1:1 coground with microcrystalline cellulose and the corresponding physical mixture are shown in Figure 1. The percent of drug dissolved in the first 20 minutes (%D20`) as a model independent parameter was employed to compare the profiles. The values of %D20` for unground drug powder, physical mixture, ground drug powder and drug coground with microcrystalline cellulose were 40, 49, 71, and 97.5, respectively.
X-ray crystallography, IR spectroscopy and Differential scanning calorimetery
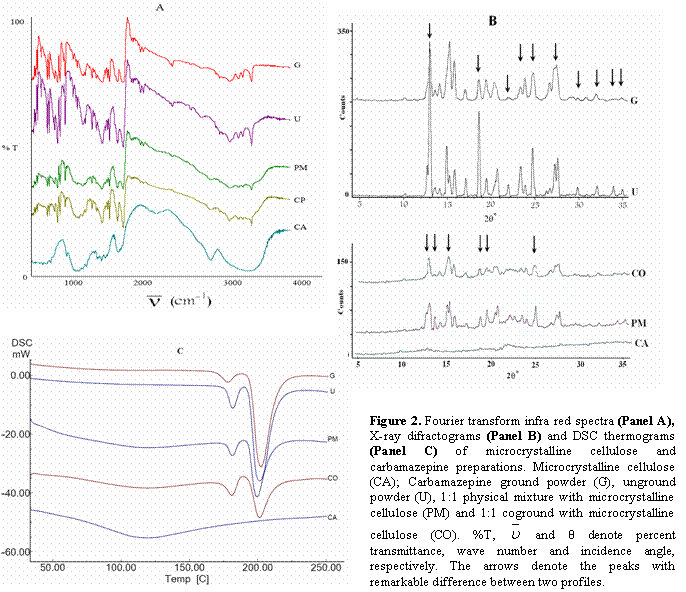
The unground carbamazepine powder showed an x-ray pattern identical to that reported for β (III) polymorph (22, 23) with characteristic diffraction peaks at: 2°θ values of 14.9, 15.2, 15.8, 27.2, 27.5, and 32.0°. However the ground drug showed a slightly reduced crystallinity as seen from less intensities for some characteristic peaks (Figure 2)
Comparing FT-IR spectrum of unground with that of ground carbamazepine (Figure 2) revealed no distinctive changes after vigorous grinding in a ball mill. Both unground and ground carbamazepine powders exhibited identical FT-IR spectra with absorption bands characteristic of polymorph III (22) at 3464 cm-1 (-N-H stretching), 1677 cm-1 (-C=O stretching), 1605 and 1593 cm-1 (range of -C=C- and –C=O vibration and -NH deformation), 1383 cm-1, 1271 cm-1 (-C≡N bond), 1245 cm-1 and 1019 cm-1.
Solid-solid transition from polymorph III to polymorph I upon heating has been reported for carbamazepine (9, 22). DSC thermograms of unground and ground carbamazepine (Figure 2) showed two endotherms of fusion. The first peak corresponded to the melting of form III (167.78 °C), followed by crystallization and melting of polymorph I (193.42°C). This behavior is typical for polymorph III (9, 22).
The coground system showed more reduced crystallinity. This was evident from smaller characteristic peaks in the x-ray diffractogram and smaller corresponding endotherm in DSC thermogram as compared to those of the corresponding physical mixture. But no difference was observed in FT-IR spectra indicating no chemical interaction between the drug and the carrier (Figure 2).
In vivo studies
The arithmetic mean serum concentrations of carbamazepine versus time for unground drug powder and the 1:1 coground system are seen in Figure 3. The mean values of the main pharmacokinetic parameters i.e. maximum drug serum concentration (Cmax), time to reach Cmax (Tmax) and area under the serum drug concentration between times 0 and 16 hr, ![]() , were significantly different from the corresponding values of the unground drug powder as inferred from the paired t-test analyses (p<0.0005 in all cases). The mean values of the mentioned parameters for coground system and unground powder, respectively, were: Cmax, 15.76±0.56 and 5.89±0.71 μg/ml; Tmax, 3.20±0.20 and 6.09±1.31 hr;
, were significantly different from the corresponding values of the unground drug powder as inferred from the paired t-test analyses (p<0.0005 in all cases). The mean values of the mentioned parameters for coground system and unground powder, respectively, were: Cmax, 15.76±0.56 and 5.89±0.71 μg/ml; Tmax, 3.20±0.20 and 6.09±1.31 hr; ![]() , 118.96±4.98 and 54.17±10.34 μg.hr/ml.
, 118.96±4.98 and 54.17±10.34 μg.hr/ml.
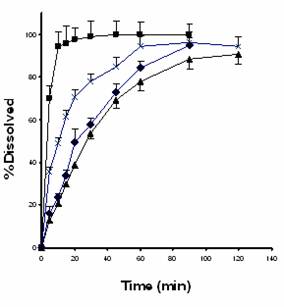
Figure 1. Dissolution profiles of carbamazepine from unground drug powder (▲), 1:1 drug:microcrystalline cellulose physical mixture (¨), ground drug powder (´) and 1:1 drug:microcrystalline cellulosecoground system (■). Each point is average of three replications. Vertical bars represent standard error of mean.
In vitro-in vivo correlation
The double reciprocal area method resulted in the following relationship between in vitro and in vivo parameters:
![]() (3)
(3)
R2=0.9993, N=20, p < 0.0001, F=25925.62
The last sampling times for in vivo and in vitro were 16 hrs and 15 minutes, respectively. The choice of 15 min was based on the fact that the dissolution of coground was nearly complete at this time and a better result was obtained when the last sampling times were equal i.e. 15 min for all dissolution profiles. Also nearly identical results were obtained if 10 or 20 min would be taken as the
last sampling times. The representative plot of equation 3 is seen in Figure 4. R2 is coefficient of determination, N is the number of data, p is probability level and F is Fisher value.
Table 1. Mean times (± standard deviation) of various strychnine induced events and percent mortality 3 and 6 hours following 0, 2 and 2.5 mg peroral doses of carbamazepine unground powder and coground. |
|||||
Event |
Control (Dose=0) |
Unground powder (2mg) |
Coground (2 mg) |
||
3 hr |
6 hr |
3 hr |
6 hr |
||
139.6 (±33.2) |
162.1(±34.6) |
* |
** †† |
** † |
|
Onset of tonic seizures (sec) |
215.6(±27.4) |
241.1(±49.5) |
262.7(±40.9) |
** †† |
** † |
Mortality percent |
100 |
100 |
80 |
** †† |
** † |
Event |
Control (Dose=0) |
Unground powder (2.5 mg) |
Coground (2.5 mg) |
||
Initial latency time (sec) |
139.6 (±33.2) |
3 hr |
6 hr |
3 hr |
6 hr |
158.1(±40.1) |
** |
** †† |
** |
||
Onset of tonic seizures (sec) |
215.6(±27.4) |
221.2(±44.3) |
** |
** †† |
** †† |
Mortality percent |
100 |
100 |
* |
** †† |
* † |
Asterisk denotes statistically significant difference between mean values of preparations (unground and coground) and control. * indicates p<0.05 and ** indicates p<0.01. Obelisk denotes statistically significant difference between mean values of unground and coground at corresponding indicated times. † indicates p<0.05 and †† indicates p<0.01. All values are the average of ten measurements. |
|||||
Anticonvulsive effect
The mean values of times of different strychnine induced events following 3 and 6 hr after unground drug powder and the coground administration to 10 mice as well as the control group are given in Table 1. The mean values of duration of tonic hind limb extension following the same administration schedule as above in maximal electroshock test are seen in Table 2.
discussion
As is evident from the values of %D20’ and Figure 1, the dissolution of carbamazepine from its preparations varies according to the order unground drug powder<physical mixture<ground drug powder<drug coground with microcrystalline cellulose. The higher dissolution rate of physical mixture relative to unground powder is probably due to adsorption of the hydrophilic colloidal particles of microcrystalline cellulose onto the hydrophobic carbamazepine particles, which in turn might augment the wettability and deaggregation of the latter particles (24-29).
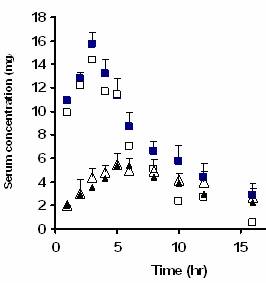
Figure 3. Observed carbamazepine serum concentration versus time plots after administration of 1:1 drug:microcrystalline cellulose coground system (solid squares) and unground drug powder (solid triangles). Each point represents the average of 11 rabbits. Vertical lines represent the standard errors. Predicted serum concentrations obtained from the in vitro in vivo correlation are shown by open squares for the coground system and by open triangle for the unground powder.
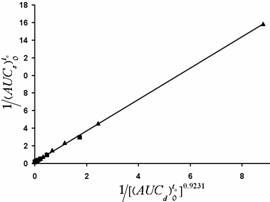
Figure 4. Double reciprocal plot of ![]() against
against ![]() indicating a direct in vitro in vivo correlation for unground carbamazepine powder (▲) and 1:1 drug:microcrystalline cellulose coground (■).
indicating a direct in vitro in vivo correlation for unground carbamazepine powder (▲) and 1:1 drug:microcrystalline cellulose coground (■).
The increased dissolution of ground drug with respect to the unground drug powder and physical mixture can be attributed to the reduced crystallinity characterized by less intense peaks in the x-ray diffraction pattern (Figure 2), reduction of drug particle size and hence enhanced effective surface area under the intense milling conditions.
The coground system exhibits the highest dissolution rate due to more reduction in the drug crystallinity characterized by less intense peaks in the X-ray diffraction and smaller endotherm in DSC thermogram (Figure 2), enhanced wetability in the presence of the carrier and particle size reduction i.e. increased effective surface area during the preparation.
For the reasons mentioned above the coground system dissolves efficiently at the absorption site during the residence time which enhances both rate of drug bioavailability as well as its apparent extent of bioavailability. This enhancement is clearly evident from the main pharmacokinetic parameters i.e. higher Cmax, shorter Tmax and higher ![]() of the coground as compared with those of unground drug powder. It can be seen from equation 2 and Figure 4 that there is an excellent novel point by point (a kind of level A) correlation between in vitro dissolution and in vivo data. This approach is a kind of simple deconvolution technique which unlike most other deconvolution methods requires no intravenous data.
of the coground as compared with those of unground drug powder. It can be seen from equation 2 and Figure 4 that there is an excellent novel point by point (a kind of level A) correlation between in vitro dissolution and in vivo data. This approach is a kind of simple deconvolution technique which unlike most other deconvolution methods requires no intravenous data.
In the deconvolution methods leading to traditional level A correlation there is a linear relationship between fraction of drug absorbed up to any time and percent drug dissolved in vitro up to the same time. The present novel IVIVC uses the partial area under the curve, AUCs instead of the fraction absorbed and its correlation with dissolution is non linear (equation 1). In the case of class II drugs, AUCs depends highly on the fraction of drug absorbed which in turn is influenced by the dissolution. Also AUCs is affected by drug clearance from the body. Due to profound effect of dissolution and/or fraction absorbed it is expected that AUCs relates to dissolution, but due to the interference of the opposing effect of clearance the in vitro-in vivo relationship is non-linear. The APD of the novel IVIVC (equation 3) is 8.71 for the values of partial areas which is quite acceptable considering the inherent error associated with integration process employing the trapezoidal rule.
Table 2. The mean values (± standard deviation) in seconds of duration of tonic hind limb extension 3 and 6 hours after administration of 0, 2 and 2.5 mg peroral doses of carbamazepine unground powder and coground. |
||||
Control (Dose=0) |
Unground powder (2 mg) |
Coground (2 mg) |
||
3 hr |
6 hr |
3 hr |
6 hr |
|
19.2 (±4.49) |
17.9 (±2.3) |
** |
** †† |
** †† |
Control (Dose=0) |
Unground powder (2.5 mg) |
Coground (2.5 mg) |
||
3 hr |
6 hr |
3 hr |
6 hr |
|
19.2 (±4.49) |
17.6 (±2.5) |
** |
** †† |
** †† |
Asterisk denotes statistically significant difference between mean values of preparations (unground and coground) and control. ** indicates p<0.01. Obelisk denotes statistically significant difference between mean values of unground and coground at corresponding indicated times. †† indicates p<0.01. All values are the average of ten measurements. |
||||
In order to carry out the prediction of the drug serum concentration from the IVIVC, the experimental ![]() is inserted in equation 3 to compute
is inserted in equation 3 to compute ![]() and then the drug serum concentration is calculated as follows:
and then the drug serum concentration is calculated as follows:
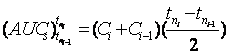 (4)
(4)
Where Ci and Ci-1 are drug serum concentrations at times tni and tni-1. When tni-1=0, C1 can be calculated from equation 4. When tni=1, tni-1 equals zero therefore Ci-1 becomes zero and Ci=C1 then C1 is calculated by:
 (5)
(5)
And generally:
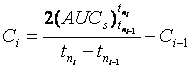 (6)
(6)
In which
![]()
The predicted serum concentrations against time are given in Figure 3 and the accuracy of prediction is seen from the linear plot of observed serum concentrations (Cs,obs) versus predicted ones (Cs,pred) (Figure 5). The linear correlation is described by: Cs,obs= 1.0152 Cs,pred + 0.5065 (7) R2=0.9194, N=20, p < 0.0001, F=205.6 The average percent deviation (APD) for the predicted drug serum concentration is 18.61.
The results of anticonvulsive investigations are also in parallel with those of serum concentration studies. As can be seen from Tables 1 and 2 the protective effect of coground system against seizure events in comparison with that of unground drug powder is considerable and depends on dose and time of carbamazepine administration. For example the protection effect of coground system is between 40-140 % higher than that of unground powder. Furthermore the unground drug powder is also effective although it is not as much as the coground system (Tables 1 and 2).
In conclusion, the cogrinding technique is an efficient method for increasing carbamazepine dissolution, serum concentration and anticonvulsant effect. Cogrinding may be superior to other techniques of solid dispersion preparation in that it does not involve toxic solvents (6) and sophisticated equipments (9). In addition the double reciprocal area approach presented in this study is a simple point by point (a kind of level A) correlation, which is superior to most other deconvolution methods since it requires no intravenous data. Such an approach has already been applied successfully to glibenclamide (25). Thus it seems that this approach is a promising deconvolution method not only for carbamazepine preparations but also may be extended for other drugs exhibiting dissolution rate-limited bioavailability.
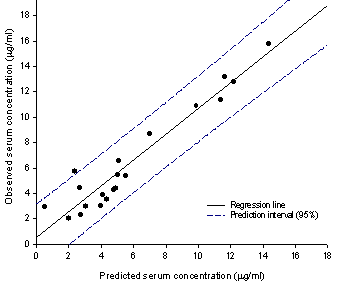
Figure 5. Observed carbamazepine serum concentrations versus predicted values based on proposed novel in vitro in vivo correlation. The upper and lower 95% prediction interval is represented by the broken lines.
Acknowledgements
This work was supported by a joint grant from Research vice Chancellery and Drug Applied Research Centre, Tabriz University of Medical sciences.