Transbuccal Delivery of Acyclovir (II): Feasibility, System Design, and In Vitro Permeation Studies Amir
H. Shojaei1, Sen-lin Zhou2,
Xiaoling Li3 Abstract Purpose. To design a buccal mucoadhesive system for systemic delivery of acyclovir using a novel mucoadhesive, copolymers of acrylic acid and poly(ethylene glycol), and to determine the feasibility of transbuccal delivery of acyclovir using this system. Methods. The buccal delivery system was prepared using an adhesive, a copolymer of acrylic acid and poly(ethylene glycol) monomethylether monomethacrylate, and an impermeable membrane to prevent excessive washout by saliva and to attain unidirectional release. Acyclovir was loaded into the copolymer film prior to lamination of backing material. In vitro drug release studies were conducted in isotonic McIlvaine buffer solution. Buccal permeation of acyclovir was investigated using porcine buccal mucosa with side-by-side flow through diffusion cells at 37°C. Acyclovir was quantified using HPLC. Results. Buccal permeation of acyclovir from the mucoadhesive delivery system was controlled for up to 20 hours with a time lag (tlag) of 10.4 hours and a steady state flux of 144.2 µg/cm2/h. With the incorporation of NaGC into the system tlag was shortened to 5.6 hours with an enhanced steady state flux of 758.7 µg/cm2/h. Sustained delivery of acyclovir across bucccal mucosa using this mucoadhesive system was maintained for up to 22 hours. Conclusions. The mucoadhesive system of P(AA-co-PEG) was shown to be a good candidate for controlled oral mucosal delivery of acyclovir. Buccal delivery of acyclovir was proven feasible based on in vitro permeation studies. Introduction Acyclovir is one of the most effective and selective agents against herpes viruses. It is widely used to treat herpes simplex virus (HSV) infections in both immunocompetent (1) and immunocompromised (2) patients. Current available dosage forms of acyclovir are intended for intravenous, oral, and topical administration. Systemic delivery of the drug following administration by these routes is far from being optimal, however. The oral absorption of acyclovir is dose dependent and highly variable with bioavailability ranging from 15 to 30% (3). The percutaneous penetration of acyclovir is poor (4). The limited solubility of acyclovir in water makes intramuscular administration rather difficult. Strongly alkaline (pH 10-11) solutions are required for intravenous infusion or bolus, which may result in thrombophlebitis or perivascular inflammation. There have been many attempts documented in the literature for improving the physico-chemical properties of acyclovir by chemical modification. These include using highly water soluble esters of alkylamines and benzylcarbamates derivatives of acyclovir as prodrugs (5) for ocular or parenteral formulations and aliphatic prodrugs of acyclovir (6) to enhance its nasal transport, employing novel redox-based chemical targeting systems (7) for improving intradermal delivery, as well as utilizing 6-deoxyacyclovir prodrug (8) and 1-valyl ester prodrug, valacyclovir (9), for greater oral bioavailability. However, a logical approach to circumvent the bioavailability and physico-chemical problems of acyclovir is to deliver the drug through an alternative route.Absorption of drugs through the oral cavity was noted as early as 1847 by Sobrero, the discoverer of nitroglycerin, and systemic studies of oral cavity absorption were first reported by Walton and Lacey in 1935 (10). Since then, substantial effort has been focused on drug absorption from a drug delivery system in a particular region of the oral cavity (11, 12, 13, 14). As a site for drug delivery, the oral cavity offers many advantages over other routes of drug administration. The mucosal lining of the oral cavity are readily accessible (15), robust, and heal rapidly after local stress or damage (15, 16, 17). Oral mucosal drug delivery systems can be localized easily and are well accepted by patients (18). Therefore, it is evident that the oral cavity can serve as a site for systemic drug delivery. The total surface area of the oral cavity is about 100 cm 2 (19). The mucosal membranes of the oral cavity can be divided into five regions: the floor of the mouth (sublingual), the buccal mucosa (cheeks), the gums (gingiva), the palatal mucosa, and the lining of the lips. These oral mucosal regions are different from each other in terms of anatomy, permeability to drug, and their ability to retain a system for a desired length of time. Although the buccal mucosa is less permeable than the sublingual mucosa and it does not yield a rapid onset of action as seen with sublingual delivery, mucosa of the buccal area has an expanse of smooth and relatively immobile surface, which is suitable for placement of a retentive system. These characteristics make the buccal mucosa a more appropriate site for prolonged systemic delivery of drugs. It has been shown that buccal route offers excellent opportunities for systemic delivery of drugs. In general, drug delivery through this route has the advantages of preventing the drug from degradation in the gastrointestinal tract, avoiding first-pass effect, and bypassing gastrointestinal absorption. The latter advantage is the rationale for designing a transbuccal drug delivery system of acyclovir to overcome the low oral bioavailability of the drug.The purposes of this study were to design a buccal delivery system for acyclovir using a novel buccal adhesive and to evaluate the feasibility of transbuccal delivery of acyclovir through in vitro release and permeation studies using porcine buccal mucosa. The novel mucoadhesive hydrogels of copolymer [P(AA-co-PEG)] of acrylic acid (AA) and poly(ethylene glycol) monomethylether monomethacrylate (PEGMM) (20) were used in designing the transbuccal delivery system. Materials And Methods Preparation of the Polymeric Films Mucoadhesive films were prepared by radical copolymerization in a mold as described elsewhere (20). Briefly, acrylic acid (AA) (Aldrich Chem. Co., Milwaukee, WI), polyethylene glycol (PEG molecular weight of 400) monomethylether monomethacrylate macromers (PEGMM), and ethylene glycol dimethacrylate (EGDMA) (Polysciences, Inc., Warrington, PA) were dehibited for 24 hours using De-hibit 100 ion exchange resin (Polysciences, Inc., Warrington, PA). Appropriate amounts of 2,2,-azobisisobutyronitrile (AIBN) (Janssen Chemica, Belgium) were dissolved in AA and PEGMM (M/I = 1000) with varying amounts (0 to 1.3 wt%) of EGDMA and purged with nitrogen. The solutions were then degassed and filled into molds constructed with two glass plates using silicon rod as a spacer. Copolymerization was carried out at 80°C for 18 hours. The resulting films were placed in tetrahydrofuran (THF) (24 hours) followed by methanol (16 hours) and de-ionized water (24 hours). Solvents were constantly changed with fresh solvent to remove unreacted monomer and initiator. Assessment of Mucoadhesive Force The force detection system (20) consisted of a precision load cell (GS-500, Transducer Techniques, Temecula, CA) with a hook attachment. Analog signals generated by the load cell were converted into digital signals by an A/D converter (Model 500A, Keithley Metrabyte, Taunton, MA). Data were recorded using EasyLX software (Keithley Metrabyte) and an IBM compatible computer. Polymer films, 0.5 cm in diameter, were glued onto glass slides (Labsyn) using cyanoacrylate adhesive. The films were pre-hydrated with the test medium, isotonic McIlvaine buffer, at 37°C for 5 minutes. They were then brought in contact with porcine buccal mucosa by the weight of the sample holder (8.6 g) for 1 minute. The platform attached to the load cell transducer was raised at a constant speed of 50 µm/s by a precision geared motor and the force required for the fracture of mucoadhesive bond was recorded. Five films of each composition were assessed in the force measurement studies. Labsyn 3 x 1 cm pre-cleaned plain glass slides were used as blank controls. Poly(acrylic acid) crosslinked with 0.3 wt% EGDMA (cr-PAA) films were used as positive control because of their known bioadhesive properties (21). Determination of Glass Transition Temperature The glass transition temperature (Tg) of the copolymeric film was determined using a Shimadzu differential scanning calorimeter (DSC-50) and a Shimadzu thermal analyzer (TA-50) data system. The measurements (4-6 mg samples) were performed with a heating rate of 10°C/min to a maximum temperature of 220°C. Tg measurements were conducted in triplicates. Analytical Method HPLC system consisted of a Waters M590 pump (Waters, Milford, MA), a Gilson 115-UV absorbance detector (Gilson, Middleton, WI), a Hitachi L-7200 autosampler (Hitachi, Tokyo, Japan), a reverse phase Beckman C-18 column (Ultrasphere, 5 m m, 4.6 mm x 15 cm), and EZChrom software (Scientific Software, CA). A mobile phase composed of 0.02M K 2HPO4: Methanol (90:10) was used for eluting the drug. Acyclovir was detected at UV wavelength of 250 nm.Drug Loading and In Vitro Release Studies Acyclovir (Sigma Chemical Co., St. Louis, MO) was loaded into the buccal adhesive by equilibrium swelling of the copolymeric films in isotonic McIlvaine buffer (pH 6.8) solutions of the drug at 37°C for 24 hours. Salt content was determined by hydrating the films in isotonic McIlvaine buffer solutions (without acyclovir). The permeation enhancer was incorporated into the system by equilibrium swelling of the copolymeric films in a 2.58 mg/ml solution of acyclovir containing 2 mM NaGC in isotonic McIlvaine buffer (pH 6.8). Percent drug loading was then calculated according to the following expression where Dry Weight Loaded denotes the dry weight of the film after equilibrium swelling in isotonic McIlvaine buffer solutions of acyclovir and Dry WeightNon-hydrated denotes the dry weight of the film before swelling.The distribution of acyclovir between the film and the loading solution was determined from the slope of a plot of drug in the film vs. drug in loading solution. The drug release studies were conducted by placing the films in flow through chambers filled with isotonic McIlvaine buffer (pH 6.8) (simulated gingival fluid) at 37°C. The flow rate of release medium was controlled by a peristaltic pump (Minipuls 3, Gilson, Middleton, WI) at 2 mL/15 minutes for the first 4 hours followed by 2 mL/hr for 8 hours. The samples were collected by using a Gilson-203 fraction collector and analyzed for acyclovir content using HPLC. Release rates were calculated by dividing the slope of cumulative amount released versus time curve and the surface area of the film. Buccal Tissue Preparation Porcine buccal tissue (Long Ranch, Inc., Manteca, CA) was stored in Krebs buffer at 4°C and used within 2 hours after slaughter. The buccal mucosal membranes were separated by removing the underlying connective tissues using surgical scissors. Buccal mucosal membranes with an approximate area of 0.75 cm 2 were then mounted between the donor and the receiver chambers of diffusion cells.Transbuccal Permeation Studies Side-by-side flow-through diffusion cells (Crown Glass Co., NJ) with a diffusional area of 0.69 cm 2 were used. Temperature was maintained at 37ºC by water jackets surrounding the two chambers. Both chambers were stirred with Teflon coated magnetic stirring bars. After buccal membranes were equilibrated with Krebs buffer (310 mOsm) in both chambers, the receiver chamber was filled with the fresh Krebs buffer and the donor chamber was charged with acyclovir solution (2.4 mg/mL). The flow rate of buffer was controlled at 1.23 mL/hr with a peristaltic pump. The samples (n=3) were collected every 90 minutes by using a Gilson-203 fraction collector and analyzed using HPLC method for acyclovir content. Buccal tissue viability in terms of permeation was verified by using the same buccal tissues to conduct two consecutive permeation experiments. Both sets of experiments were run under identical settings with a 4-hour equilibration/washing period between the first and the second experiment. The permeability coefficients (P) were calculated as follows:where dQ/dt is permeation rate, C is the concentration of the donor chambers, and A is the surface area of diffusion. Experimental fluxes were determined by dividing the slope of cumulative amount permeated vs. time curve by the diffusional area.To investigate the unidirectional drug delivery, buccal permeation of acyclovir from the system was investigated by applying the mucoadhesive films backed by impermeable membranes (R-Coat, 2-EST-A-S242M, Release International, West Chicago, IL) onto the buccal mucosa mounted between side-by-side flow through diffusion cells. The temperature was maintained at 37°C. The flow rate of buffer for receiver chamber was controlled at 1.23 mL/hr with a peristaltic pump. The samples (n=3) were collected every 90 minutes by using a Gilson-203 fraction collector. Samples were analyzed for acyclovir content using HPLC. Results And Discussion It is currently believed that the permeability barrier in the oral mucosa is a result of intercellular material derived from the so-called ‘membrane coating granules’ (MCG) (22, 23, 24). Because of the similarity to the human non-keratinized buccal epithelium, pig is found to be a good model for studying buccal permeation of drugs (25). In the present study, the permeability coefficients (6.21±0.60 x 10-6 and 6.09±0.71 x 10-6 cm/sec) and the t lag values from tissue viability experiments were determined and no significant difference (P>0.05) was found. The results indicated that permeability barrier was not altered within the time span of study (28 hours).The results from our previous studies (26) on solution permeation of acyclovir across porcine buccal mcuosa revealed an excellent linearity (r2 = 0.993) between steady state flux and donor chamber solution concentration of acyclovir. It was thus concluded that solution permeation of the drug was through a passive diffusion process over the investigated range (0.8-2.4 mg/mL). Permeation enhancement studies conducted using the trihydroxy bile salt, sodium glycocholate (NaGC), showed that co-administration of NaGC increased the steady state flux of acyclovir through porcine buccal mucosa from 2 to 9 times. The enhancement ratio (ER) increased steadily with increasing NaGC concentration up to 20 mM, beyond this point ER leveled off to a plateau (26). Based on the permeation enhancement in the solution transportation experiments, an acyclovir buccal delivery system was designed by incorporating NaGC in the system. Novel copolymers of acrylic acid and PEGMM were designed to optimize mucoadhesion. The copolymer films with an AA:PEGMM mole ratio of 84:16 were shown to afford a favorable thermodynamic profile for mucoadhesion and have the highest mucoadhesive force . The mucoadhesive performance of the P(AA-co-PEG) films was compared to that of a known mucoadhesive, cr-PAA. The force of buccal mucoadhesion of the copolymer films containing 16 mole % PEGMM was 39% higher (P<0.05) than that measured from cr-PAA films. The glass transition temperature of the drug-loaded copolymeric film (with 7.26% drug loading) was 34.1±3.8°C in the non-hydrated state, which makes it possible to apply an elastomer device in dry state by warming the device. The effects of drug content on the force of adhesion of bioadhesives have been reported. Anlar et al. (27) found that drug content (morphine sulfate, 15 wt%, MW 669) significantly decreased mucoadhesive force in the hydroxypropyl methylcellulose-carbopol buccoadhesive tablets, whereas Ponchel et al. (28) reported no significant reduction in the bioadhesive bond strength (measured as work of adhesion) due to drug content (metronidazole, 50 wt%, MW 171) in poly(acrylic acid)-hydroxypropyl methylcellulose tablets. In this study, the effect of drug content on mucoadhesive force was determined using the 1.3 wt% EGDMA containing films loaded with different amounts of acyclovir. The presence of acyclovir (0.56% - 7.26%) did not significantly (P>0.05) affect the force of mucoadhesion (Table 1).Because the effect of drug content on mucoadhesive force could be related to the size and the hydrophilicity of the drug as well as potential interaction between drug and mucoadhesive, the result of the effect depends on the physical chemical properties of the drug and the adhesive. When hydrophobic drugs are loaded in hydrophilic matrices, drug content may significantly affect mucoadhesive strength by changing the surface properties of the bioadhesive thus retarding or enhancing the formation of an intimate contact between the buccal and the adhesive surfaces. Table 1 Effect of drug content on mucoadhesive force of P(AA-co-PEG) films containing 1.3 wt% EGDMA
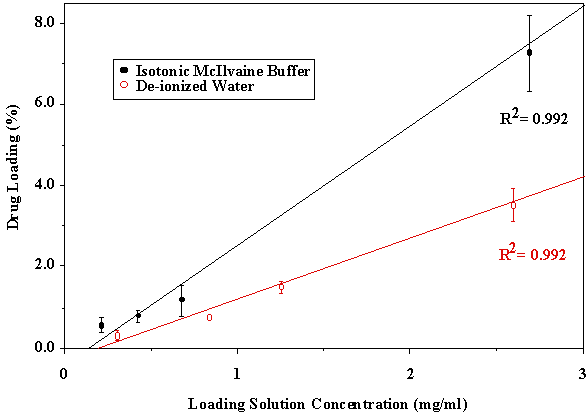
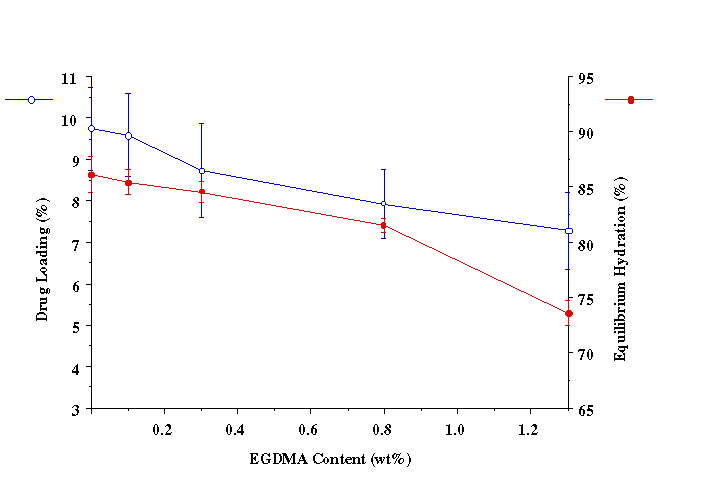
ANOVA P>0.05 When intermediate to large molecular weight (>500) drugs are loaded in bioadhesive polymeric matrices, the drug may act as a physical crosslinker. Thus, chain mobility and the swelling rate of the polymer may decrease. Both of these factors could cause a decrease in the mucoadhesive bond strength, especially when drug loading content is relatively high. Since acyclovir was completely released from the P(AA-co-PEG) film, no interaction between drug and polymer could cause the decrease in chain mobility and swelling. Therefore, no significant effect of drug loading on mucoadhesion was observed. Drug loading was found linearly related to the concentration of drug in loading solution (Figure 1). Since the drug was loaded by equilibrium swelling, the factors affecting hydration altered drug loading. Equilibrium hydration was influenced by the crosslinking density and the properties of loading solution. Increasing crosslinking density decreased drug loading (Figure 2). Drug loading was doubled as the loading solution medium was changed from de-ionized water (DIW) to isotonic McIlvaine buffer (pH 6.8). This was attributed to the increase in hydration of the films in isotonic McIlvaine buffer as compared to DIW. The enhanced hydration is due to the higher pH of the buffer than that of DIW. The higher the pH of the swelling medium, the greater the degree of ionization, which in turn causes the polymeric network to expand because of electrostatic repulsion. Figure 1. Effects of loading solution concentration and medium on drug loading at 37°C (Film Composition: 84:16 mole% AA:PEGMM with 1.3 wt% EGDMA). Figure 2. Effect of EGDMA content on equilibrium hydration and drug loading at 37°C. The P(AA-co-PEG) films containing 16 mole% PEGMM crosslinked with 0.1, 0.3, 0.8, 1.3 wt% EGDMA were prepared to prolong the release of acyclovir. Films containing 1.3 wt% EGDMA (Table 2) sustained the release for up to 4 hours. Although the increase in crosslinking density would prolong the release time, the increased crosslinking density may cause a decrease in adhesion. It has been shown that crosslinking density could influence mucoadhesive performance of PAA by changing the effective number of PAA chains in a given volume (chain density) and the mobility of PAA chains. Table 2. Effects of EGDMA on the in vitro release of acyclovir and the force of mucoadhesion
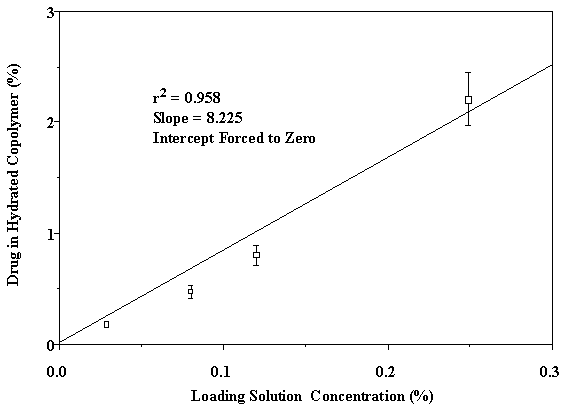
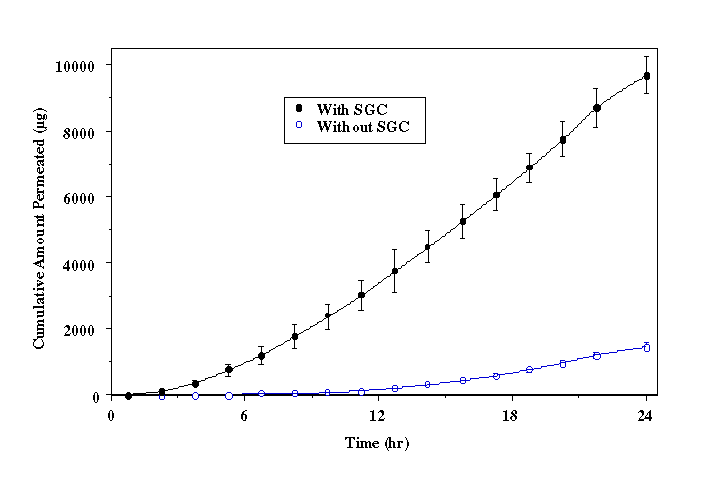
† Blank Control: Labsyn pre-cleaned plain cut 3"x1" slides, Mucoadhesive Force = 0.74 ± 0.11 N/cm2. Park and Robinson (21) demonstrated that mucoadhesion of PAA hydrogels decreases as the concentration of crosslinking agent is increased. Introduction of a crosslinker results in a higher Tg, which decreases mucoadhesion due to reduction of chain mobility and interpenetration (29). However, the mucoadhesive force of the EGDMA crosslinked P(AA-co-PEG) films showed no significant (P>0.05) adhesion loss (Table 2). This may be attributed to the contribution of PEG in the copolymer. As shown in our previous publication (20), PEG in the copolymer is able to form hydrogen bond with mucosal surface. Increase in crosslinking density causes lower swelling, which would, in-turn, result in a higher number of PEG groups in a given surface area for mucoadhesion. Therefore, decreased chain flexibility is compensated by increased PEG group density, which facilitates the formation of hydrogen bonding. To further understand the transportation of acyclovir from the copolymeric system across the buccal mucosa, the relationship between the delivery system and the surrounding liquid phase was analyzed. The equilibrium hydration of the films was used to determine the distribution of acyclovir between loading solution and the hydrated copolymer film. As shown in Figure 3, acyclovir is 8.2 times more likely to be in the film than in the loading solution. By using the distribution coefficient, the surface area and the thickness of the film at equilibrium hydration, the concentration of acyclovir in the film can be converted to that in solution at equilibrium state. In the present study, the aqueous phase that lies between the mucoadhesive device and the surface of buccal mucosa can be considered as the solution. Figure 3. Distribution of acyclovir between P(AA-co-PEG) and loading solution. Based on the solution permeation studies , the steady state flux of acyclovir using the mucoadhisive system loaded with 9.8 ± 1.2 mg of acyclovir is predicted as 135.7±7.5 µg/cm2/h. Given that the actual experimental flux determined from buccal permeation of acyclovir from the system was 144.2 ± 8.2 µg/cm2/h, the buccal drug transport from the mucoadhesive system is also a passive diffusion process. Therefore, the delivery system does not affect the transport process.Delivery of acyclovir using the system yielded a steady state flux of 144.2 ± 8.2 µg/cm2/h with a tlag of 10.5 hours (Figure 4 without enhancer). However, based on the effective inhibitory concentrations (30-500 µg/L) against HSV-1 and HSV-2 and the mean total body clearance (15.6 L/h) of acyclovir (30), the target steady state flux (Jss) for delivering acyclovir through the buccal mucosa is estimated to be 0.468 to 7.8 mg/h for a normal adult using the following equation: where Cpss represents the effective inhibitory concentration and Cl is the total body clearance. Figure 4. In vitro buccal permeation of acyclovir from P(AA-co-PEG) mucoadhesive devices containing 7.26% of acyclovir (Film Composition: 84:16 mole% AA:PEGMM with 1.3 wt% EGDMA). To increase the flux and decrease the lag time associated with acyclovir permeation across buccal mucosa from the system, NaGC was incorporated. With the incorporation of NaGC, steady state flux increased to 758.7±50.9 µg/cm2/h and the t lag was reduced to 5.6 hours (Figure 4). The rate of drug delivery across buccal mucosa was 1.15 mg/h for a system with a surface area of 1.52 ± 0.11 cm2, which falls in the target range. And the amount of acyclovir (9.8 ± 1.2 mg) loaded into this system is within the desired range. Therefore, transbuccal controlled release delivery of acyclovir is feasible according to the calculated target flux and the in vitro permeation studies.Conclusion A new mucoadhesive system for the buccal delivery of acyclovir was developed using the copolymer of acrylic acid and poly ethylene glycol monomethylether monomethacrylate. The presence of acyclovir did not affect the adhesion of P(AA-co-PEG). Systemic delivery of acyclovir from the mucoadhesive delivery system through the buccal mucosa was found feasible based on in vitro permeation studies. Acknowledgement Authors would like to thank Dr. Bret Berner for stimulating discussions and Long Ranch, Inc., Manteca, CA for kindly supplying porcine buccal tissue. References
1Present address: Faculty of Pharmacy & Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, T6G 2N8 Canada, email:ashojaei@pharmacy.ualberta.ca 2Present address: Children's Hospital, Shanghai Medical University, Shanghai, 200032, P.R. China, email:luhong@public2.sta.net.cn 3Corresponding author: Xiaoling Li, Department of Pharmaceutics and Medicinal Chemistry, School of Pharmacy and Health Sciences, University of the Pacific, 3601 Pacific Avenue, Stockton, California, 95211 USA, email:xli@uop.edu Keywords: Acyclovir; Buccal mucosal delivery; In vitro permeation; Mucoadhesive polymers
Published by the Canadian Society for Pharmaceutical Sciences. Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences. |
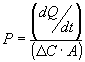
{kind=link}
{kind=link}
{kind=link}