Symposia Proceedings
CSPS Symposium, February, 1998, Ottawa Ontario A two-day Symposium was held on February 13-14, 1998 in Ottawa, Canada, to discuss the state of pharmaceutical research and development in Canada, and bioequivalence of drug products with special characteristics. The Symposium was sponsored by the Canadian Society for Pharmaceutical Sciences and the Association of Faculties of Pharmacy of Canada. Twenty six talks by 24 speakers were presented. The abstracts of the presentations are enclosed, and an overview of the talks and discussions will be forthcoming. Co-sponsor: Association of Faculties of Pharmacy of Canada Corporate Sponsors: DuPont, Searle, Biovail, Genpharm, Novartis, Phoenix, MDS Neo-Pharm Organizing Committee: Laszlo Endrenyi (University of Toronto, Ontario), Keith Gallicano (Ottawa General Hospital, Ontario), Fakhreddin Jamali (University of Alberta, Alberta), Sylvie Laganiere (Phoenix International, Montreal, Quebec), Marc LeBel (Anapharm Inc., Ste-Laurent, Quebec), Eric Ormsby (Health Protection Branch, Ottawa, Ontario), Anne Tomalin (CanReg Inc., Flameborough, Ontario)Reporters: Eugenia Palylyk-Colwell (Alberta Blue Cross, Alberta), Amir Shojaei (University of Alberta, Alberta), Okpo Eradiri (Biovail, Mississauga, Ontario) Canada, The Challenge of Becoming a Major Player in Pharmaceutical Research and DevelopmentA. Noujaim, AltaRex Inc., Edmonton, Alberta State of Pharmaceutical Research in
Canada: Present and Future Examples of Research as a Scientific
Basis of Regulation: Bioavailability Guidelines Research Discovery: Nourishing a
Platform for Growth and Competitiveness Bioequivalence and
Interchangeability: Implications for a Provincial Formulary Bioequivalence of Modified
Release Formulations: Is There a Need for a Revisit? New Standards for Bioequivalence: An
Absorption View Modulation of Drug Response by its
Rate of Input Into the Body Bioequivalence of Topical
Dosage Forms Intended for Local Action: Introduction and Overview Draft Guidance to Establish Equivalence of
Efficacy and Safety of a Second Entry Metered Dose Inhaler Evaluation of Population
Dose-Response and Relative Potency Using Mixed-Effects Models: A Rational Approach to
Compare Different Formulations of Albuterol Metered-Dose Inhalers Dermatopharmacokinetics:
Proposed use for Establishing the Bioequivalence of Topical Products Current Concepts in the Assessment of
Topical Cortisteroid Formulations for Bioquivalence Purposes Variabilities in Bioequivalence
Studies Highly Variable Drugs: Experience with
Propafenone Influence of Equilibration Diet on the
Intra-Subject Variability of a Highly Variable Drug Comparison Between Individual and
Average Bioequivalence for 3 Highly Variable Drugs Recommendations for Comparative
Bioavailability Standards For Drugs with a Narrow Therapeutic Range: Experience with
Cyclosporine Individual Bioequivalence: Attractive
Rationale, Practical Difficulties Sample Size Considerations for
Bioequivalence Studies of Narrow Therapeutic Range Drugs Narrow Therapeutic Range Drugs:
Experiences with Clozapine The Importance of Tight Manufacturing
Specifications in Narrow Therapeutic Range Drugs Is Fuzzy Logic too Fuzzy for
Bioequivalence Assessment? Bioequivalence of Chiral
Drugs: an Update Bioequivalence of
Non-Linear Drugs: Saturation of Plasma Proteins Nonlinear Kinetics and Pharmacological
Response A. Noujaim, AltaRex Inc., Edmonton, Alberta The emergence of a biopharmaceutical industry in Canada in the last 10 years has been fuelled with considerable funding from the capital markets. Small Biotech companies conducting research and development in a variety of drug development fields have been the beneficiaries of this enhanced activity, thus creating an alternative to the conventional research efforts conducted in University laboratories and large pharmaceutical companies. This trend is indicative of future profound changes in the general approach of drug discovery and funding. Canadian efforts in funding advanced biopharmaceutical research have centred around a tripartite arrangement involving Federal Government incentives, facilitation of intra and extramural research by academic institutions, as well as the emerging small biotech industries. Reorienting the thrust of pharmaceutical research in Canada will greatly impact future research programs at universities in this country. State
of Pharmaceutical Research in Canada: Present and Future Extensive pharmaceutical research is supported and carried out in Canada although it is difficult to separate out "pharmaceutical" research from other disease related research. Pharmaceutical manufacturers are now the greatest contributors to pharmaceutical research in Canada. Members of the Pharmaceutical Manufacturers’ Association contribute the most, but significant contributions are also made by the Canadian Drug Manufacturers’ Association. The incredible increase in small, research-intensive biotech companies in Canada over the past decade has created another highly significant pool of money and employment in the research sector. In contrast to this optimistic picture is the role of government and government-supported granting agencies over the same period of time. Canada ranks at the bottom of the G7 countries in terms of government support of research. The budgets for MRC and NSERC have decreased markedly while those of similar agencies in other countries have increased over the same period of time. Highly trained Canadian scientists have been forced to leave the country to find employment and suitable working conditions. MRC officials have stated that the current shortfall in research funding in Canada is $80M which will rise to $200M in the near future. The future for Canadian researchers does not look bright unless government recognizes the value of research and development and substantially increases support, particularly for basic research, as rapidly as possible. Examples
of Research as a Scientific Basis of Regulation: Bioavailability Guidelines The mission of the Health Canada, Drugs Programme is to assure that drugs available in Canada are safe, effective and of high quality. It will be illustrated how applied in-house pharmaceutical science research at the Bureau of Drug Research, with the biopharmaceutics/pharmacokinetics focus, supported this mission by contributing to development of guidelines and standards providing Canadian input to international standards. The author reviews the scientific information generated with many colleagues in support of guidelines in bioequivalence, standards for dissolution as well as potential interaction advice for specific drug product monographs. With the closing of this national resource (BDR), alternative arrangements will need to be made to provide public protection information in some of the areas of competence and CSPS could well have a role in this challenge. Research
Discovery: Nourishing a Platform for Growth and Competitiveness Funding for the granting councils has dropped since 1995. In 1998, real funding for the MRC, NSERC and SHRC will be lower than in 1985. As biomedical and health sciences research account for 50% to 65% of university-based research, the 13% reduction applied over 4 years is putting enormous pressure on university science and on MRC’s grants and awards programs. The approval rate of operating grants has dropped from 65% to less than 50% and from 28% to less a than 20% for renewal and new grants respectively. This occurs at a time when competing nations are continuing to increase the budgets of their federal agencies responsible for funding biomedical and health research. The plight of the MRC comes at a time when it has broadened its role to encompass the full range of health research and has initiated alliances and partnerships, with health care providers, provinces, the voluntary sector, and industry. This strategy has been successful in improving the health of Canadians and in creating economic dividends. However, basic research, our "core business," is suffering. Continued under-funding of basic research will soon use up the pool of new knowledge upon which all other research draws. Drying up the wellspring of innovation could have dire consequences on Canada’s competitiveness and economic growth. Over the past two years, the government has undertaken several important initiatives, such as permanent funding of the NCE, and creation of the Canadian Health Services Research Foundation, and of the Canada Foundation for Innovation. These are proof of a new perspective and constitute evidence that the government recognizes the importance of research. The well orchestrated cross country lobbying campaign for an increase in the core budget of the MRC has been heard loud and clear. We have reason to believe that the message will soon be heeded. Bioequivalence and Interchangeability: Implications for a
Provincial Formulary Is there a difference between bioequivalence and interchangeability? Why do provincial formulary committees review bioequivalence data - does this practice provide a safety valve in the system or is it merely duplication? These questions are often asked by opposing sectors in the pharmaceutical industry and at various levels of government. Some provinces have undertaken harmonization efforts between the provincial and federal review processes despite their different mandates. The populations covered by provincial government formularies, are, for the most part, the elderly, the indigent or the disabled, often with multiple comorbid disease. As a result, the generalizability of the results of a bioequivalence trial conducted in a sample of healthy adult males to populations that would not have met the inclusion criteria for the trial is circumspect. It can be argued that there is little evidence that substituting between products has resulted in clinically significant differences in these individuals; however, we cannot say with confidence that current post-marketing surveillance mechanisms are able to detect such differences. The challenge of designating products as interchangeable for these populations and the implications of new bioequivalence guidelines for complicated drugs from the perspective of a provincial formulary committee will be discussed. Bioequivalence of Modified Release Formulations: Is There
a Need for a Revisit? According to the North American guidelines, parity in the overall area under plasma drug concentration (AUC) and maximum plasma dug concentration (Cmax) suffice in proving bioequivalence of modified release formulations. It is assumed that the magnitude of Cmax is a reflection of the rate of absorption of the drug; the slower the rate of absorption the smaller should be the peak concentration. In addition since modified release formulations are often intended for the treatment of chronic diseases, it is decided that the time-course of the drug absorption may not be therapeutically relevant. It is realized that in an uninterrupted absorption phase, regardless of the kinetic order of the reaction, Cmax and Tmax are functions of rate constants governing absorption, distribution (a) and elimination (ß) rates. Since both a and ß are expected to be constant following administration of the reference and the test formulations, an inter-product difference in Cmax indicates a difference in Tmax and hence the absorption rate constant. This simple explanation, however, does not hold true when the process of absorption is somehow interrupted or when the absorption commences after a significant lag time. Two products may exhibit close absorption rate constants but one with and another without a lag time resulting in Cmax values of similar magnitudes but different Tmax’s. Depending upon the therapeutic indication, the time course of drug may be an important consideration even in chronic applications. For example, is well known that cardiovascular events are subject to circadian variations. The incidence of both myocardial infarction and sudden cardiac death are significantly greater during the morning than any other time of the day. These events coincide well with a surge in blood pressure just within the first three hours after awaking. The early morning rise in blood pressure can be suppressed with hypertensive drugs but the time of administration appear to influence the degree of blood pressure suppression. In light of the acknowledged chronobiological variation in pathophysiological events, and since Cmax does not necessarily reflects the time-course of drug absorption, it is suggested that, when appropriate, partial AUC be included as an additional criterion for bioequivalence assessment of modified release formulations. New
Standards for Bioequivalence: An Absorption View Pharmaceutical bioequivalence standards are one of the most important standards regulating the quality of drug products on the world markets today. These standards were debated in the seventies and eighties and codified by the mid-eighties in the United States. The standards that are currently accepted by the US FDA are based on plasma level determinations and include Cmax and AUC. Standards for immediate release products require a single dose study in normal subjects while controlled release products require fasted-fed and steady-state studies. These standards are empirical requiring an in vivo human study to establish bioequivalence. However, there are many oral drug products for which in vivo plasma levels are not influenced by the formulation. A new approach to setting drug bioequivalence standards includes determining the rate determining step for systemic availability, in particular, solubility and permeability. These properties plus a dissolution rate specification may allow for in vitro determination of bioequivalence and ensure in vivo bioequivalence. This approach is being developed by the USA FDA and incorporated into a soon to be released Biopharmaceutics Classification System (BCS) Guidance. For controlled release products the transit through the gastrointestinal tract is more complex. Consequently, classifying controlled release products as to drug release mechanisms, as well as solubility and permeability, is probably essential for establishing in vitro standards for controlled release products. This presentation will review the current status and progress towards establishing a more mechanistic basis for regulating bioequivalence of immediate and controlled release oral drug products based on the BCS. Modulation
of Drug Response by its Rate of Input Into the Body Originally, modified release formulations (MRF) were aimed to prolong the effect of a drug because of its rapid elimination. Actually, it has been demonstrated that the use of MRF can be extended to achieve two goals: firstly, to control the site of absorption and secondly, to reduce the rate of input of the drug into the body, since both characteristics can modulate the response to a drug. The control of the site of absorption of a drug can modulate its pharmacological response because it can increase the systemic availability of the drug, as may happen with erythromycin, diltiazem or R,S-verapamil, or because the delivery of the drug to the site of action is improved, as demonstrated with metronidazole, 5-aminosalicylic acid, and budenoside for the treatment of amebic dysentery and chronic ulcerative colitis. The response elicited by drugs modifying the fine equilibrium between conductance and effective volume in the cardiovascular system will trigger homeostatic reactions that will counterbalance the effect of the drug and decrease the response, and in addition will increase the incidence of undesired effects. The rate of input of drugs such as furosemide, bumetanide, nifedipine, nisoldipine, isradipine, propranolol, atenolol, metoprolol, and prazosin into the body will be a key factor to the homeostatic response, in such a way that MRF by reducing the rate of input of these drugs into the body may increase their pharmacological response. MRF can also modulate undesired responses, by avoiding the contact of the drug with the gastric mucosa, like happens with aspirin and other NSAIDs, and doxycycline. On the other hand, MRF by reducing peak plasma concentrations of a drug may also avoid side effects associated with the peak plasma concentrations, as has been demonstrated with nifedipine, verapamil, glipizide, and sedatives. Furthermore, MRF may reduce the incidence of side effects by fostering the appearance of tolerance to side effects, as described for barbiturates, diazepam, alprazolam, adinazolam and carbamazepine. Finally, MRF by reducing the rate of entry into the body appear to reduce the abuse liability of alprazolam. Bioequivalence of Topical Dosage Forms Intended for
Local Action: Introduction and Overview Semantic issues relating to the assessment of non-oral dosage forms often create confusion regarding specific clinical criteria and surrogate measures which could be monitored in order to establish safety and efficacy of such dosage forms. For example, transdermal dosage forms are specifically formulated for systemic absorption yet are intended for application to the skin. Some non-steroidal anti-inflammatory drugs have also been formulated for application to the skin but are they intended for absorption into the systemic circulation? Metered-dose inhalers deliver drug to target sites in the lung yet some percentage of the active is systemically absorbed. Similarly, nasal sprays used in the treatment of allergic rhinitis facilitate the deposition of active for local action in the nasal passage. On the other hand, some spray delivery systems, such as those containing nitroglycerin, are designed for oral administration into the buccal cavity and are intended for systemic action in anti-anginal therapy. What scientifically acceptable criteria can be readily measured and what methods can be used to assess such products? Some approaches to the assessment of bioavailability/bioequivalence of topical dosage forms intended for local action will be discussed together with an overview of the various methodologies. Draft
Guidance to Establish Equivalence of Efficacy and Safety of a Second Entry Metered Dose
Inhaler The presentation will discuss the Therapeutic Products Directorate’s (TPD) efforts in developing guidelines to establish equivalence of efficacy and safety of second entry bronchodilator and corticosteroid metered dose inhalers (MDI’s). Evaluation
of Population Dose-Response and Relative Potency Using Mixed-Effects Models: A Rational
Approach to Compare Different Formulations of Albuterol Metered-Dose Inhalers Evaluation of dose-response is a very important part of the development of new drugs. In this context, the goals are to identify a reasonable initial dose and subsequent dose increments to be used in patients and future clinical trials. Such information may be obtained from the shape and distribution of patient-specific dose-response relationships, as described by Sheiner et al. (Clin Pharmacol Ther 1989;46:63-77) and the ICH guideline on dose-response. Dose-response information is also used to compare drug products and determine relative potency (i.e. shift in dose-response relationship), as described by Finney (Statistical method in biological assay. 3rd edition, Charles Griffin & Co. Ltd, 1978). In this context, the slope/shape of the dose-response relationship is particularly important since a large difference in potency may lead to only a small change in response. These issues will be discussed using the results of a study in asthmatic patients who were administered increasing doses of albuterol from two different metered-dose inhaler formulations. Bronchodilation response was used to compare the two products. Different pharmacodynamic and statistical models were used to estimate relative potency and its confidence interval. This presentation will focus on the important factors that may affect relative potency estimation such as severity of asthma, slope and variability of the dose-response relationship and day-to-day variability in bronchodilator response of asthmatic patients. The different methods of data analysis will be compared using the results of this particular study and also using Monte Carlo simulations. The regulatory advantages of using the above approaches will also be discussed. Dermatopharmacokinetics: Proposed use for Establishing
the Bioequivalence of Topical Products Two formulations of the same active ingredient are considered bioequivalent if they produce equal concentrations of drug and active metabolites at the target site. The FDA has proposed that dermatopharmacokinetics (DPK) can be used to establish BE by measuring drug concentrations over time at the target site (skin), specifically the stratum corneum (SC). In order for DPK to become an accepted measure of BE, it must be shown that the DPK methods are validatable and verifiable. Although the technique of tapestripping has been available for some time, little has been done to establish that the method is specific, accurate, precise, and robust, and can therefore be validated according to established guidelines. The purpose of the current studies was to examine some of the parameters that may be important in the validation of the tapestripping assay, and to determine how methodological issues in this technique may affect the pharmacokinetic analysis. These studies reveal that the process of applying and removing the tape strips leads to wide intersubject and intrasubject variability in the amount of stratum corneum that is recovered. This variability in the amount of tissue recovered is due to several factors: inherent variability in individual skin type and variability in SC thickness at different anatomical sites of an given individual, inherent variability in the application and removal of the tape by different "operators", and variability related to the tape selected and environmental conditions. Of these factors, those related to the variable tissue recovery and its impact on the ability to normalize measurements, must be addressed in order to obtain the type of quality data needed to due a meaningful PK analysis. Current
Concepts in the Assessment of Topical Cortisteroid Formulations for Bioquivalence Purposes The human skin blanching (vasoconstriction) assay has been used successfully for the past three decades for the assessment of the release of corticosteroids from topical dosage forms. Application of corticosteroids to the skin produces a whitening (blanching) side effect, the degree of blanching being directly related to the potency of the drug entity or the extent of drug delivery from the compounded vehicle. The extent of induced blanching is, therefore, indicative of the clinical efficacy of the formulation. Assessment of the intensity of the induced blanching for bioequivalence determination has classically been performed by eye, a methodology that has been criticised because of the subjective nature of the estimation and the necessity for trained observers. This is in spite of the published evidence which documents the precision and reproducibility of the procedure. In an attempt to implement a more objective protocol for bioequivalence assessment of corticosteroid products, the FDA has released a Guidance document recommending the use of the Minolta chromameter. This instrument measures colour in terms of three indices: the L-scale (light-dark), the a-scale (red-green) and the b-scale (yellow-blue). Any colour can be expressed absolutely in terms of these three values. The Guidance protocol specifies the procedures to be followed in the initial pilot study to determine the experimental parameters for use in the subsequent pivotal assessment. However, there are several variables in the experimental protocol that investigators may manipulate (such as dose of formulation applied and skin contact time); a situation which is believed to reduce the robustness and reproducibility of the protocol. Moreover, the Guidance suggests the use of only the a-scale values in quantifying the blanching response, after elaborate correction of the data by subtraction of baseline and unmedicated site values. The resulting blanching response data is modelled in a suitable fashion to yield median effective dose and average responder data for the pivotal evaluation from which a determination of bioequivalence may subsequently be made. Recently there has been considerable discussion in the literature regarding the use of the chromameter for this purpose and, especially, the data handling procedures outlined in the Guidance have come under close scrutiny. The validity of the double-corrected data manipulation and modelling procedures has been assessed in several papers which indicate that the Guidance recommendations may not be ideal. Theory would suggest that this correction procedure is unnecessary since the instrument measures the absolute colour of the skin; as blanching proceeds so the colour of the skin changes and this should be quantified directly by the chromameter. Euclidean distance analysis of all three response values therefore seems appropriate. In recent presentations the FDA has also conceded that there may be a difference in the results obtained depending on the modelling procedure employed. The purpose of this presentation is to outline some of the shortcomings of the visual and Guidance protocols and to compare visually-assessed skin blanching response with chromameter data in terms of demonstrating a distinct response profile, data precision and suitability of the results for mathematical modelling. Variabilities
in Bioequivalence Studies In recent years, bioequivalence (BE) studies, in which two formulations of a drug are compared, have been based on 2-formulation, 2-period, 2-sequence cross-over designs. Log transformed concentration dependent parameters such as area under the plasma concentration versus time curve are then examined by ANOVA in which the effects in the model are typically Formulation, Sequence, Subject nested within Sequence, Period (sometimes called "Phase") and the Residual Error (sometimes called the "Subject by Formulation Interaction"). The Residual Error is made up of four components: The pharmacokinetic within-subject variance (which includes a component of analytical variability); the within-formulation (e.g. tablet to tablet) variance; the subject by formulation interaction; and unexplained random variance. For traditional average BE, the Residual Error is used in the calculation of a 90% confidence interval (90% CI) on the ratio of geometric means (GMR). The two formulations are declared BE if the 90% CI fits entirely into BE limits of 80-125%. Disadvantages of this approach are (i) the model assumes the within-test and within- reference variances are identical which is not necessarily the case, (ii) no information is provided on the pharmaceutical quality of the two products, and (iii) no separate estimation of the subject by formulation interaction is possible. In replicate designs, however, both test and reference formulations are administered twice such that the effects in the ANOVA model are typically: Formulation, Period, Subject , Subject by Formulation Interaction and the Residual Error. The Subject by Formulation Interaction occurs when there is a clear dichotomy between the test and reference means in at least a subset of the population sample. The clinical significance of the Subject by Formulation Interaction is a matter for conjecture, as is the question of how to sample the population so as to ensure representation of all relevant subsets of the population in the study sample. At present, there is no approved method of calculating CIs based on replicate designs, although a great deal of attention is being given to the concept of individual BE for which replicate designs are essential. Attention is also being given to the fact concern that fixed BE limits of 80-125% are not appropriate for all drugs. Potent drugs with narrow therapeutic indices tend to have small CIs which permit the GMR to vary a great deal from the ideal of unity (100%). On the other hand, many relatively safe drugs with high within-subject variability have very wide CIs which means that large numbers of subjects are required to give adequate statistical power, even when the GMR is close to unity. Evidence will be presented to show that scaling the BE limits to make the conditions more conservative for potent drugs with steep dose response curves reduces the risk of two generic formulations being BE with the same reference product but not BE with each other. On the other hand, broadening the BE limits for safe, highly variable drugs increases statistical power and reduces the number of subjects required. Finally we shall briefly address the question of individual BE in terms of scaled and unscaled aggregate metric that includes both means and variance terms. We conclude: (i) some form of scaling is appropriate; (ii) scaling to the within-subject variance of the reference product is problematical when the latter is a poor pharmaceutical product; (iii) scaling to a pooled estimate of variance is appropriate when the test and reference variances are close; (iv) In our experience, the individual BE metrics are elegant in concept but can be unpredictable in practice. Highly
Variable Drugs: Experience with Propafenone Propafenone is an antiarrhythmic agent which is primarily used for the treatment of documented life-threatening ventricular arrhythmias, such as sustained ventricular tachycardia. Following oral administration, it is nearly completely absorbed but undergoes extensive first-pass hepatic metabolism. A comparative bioavailability study was conducted to determine the bioequivalence of a generic formulation of propafenone tablet to the Canadian reference product, Rythmol, under fasting conditions. Eighteen normal, healthy male volunteers entered and completed the study. A single-dose (1 x 300 mg tablet) standard randomized, two-way crossover design with a washout period of 1 week was used for the study. Plasma levels of propafenone after the oral administration of the two products were measured by a validated HPLC/UV method (LOQ = 5.0 ng/mL). The results of the study can be summarized in the following table:
The statistical analysis (ANOVA) of the data revealed no significant differences (p>0.05) in any of the parameters between the two products. However, the T/R ratio of the means was well over 110% with the 90% confidence interval (CI) outside the 80-125% interval for both AUCT and Cmax. Using current TPD criteria the two products would not be considered bioequivalent. A detailed examination of the in-vitro dissolution data showed that the two products provided very similar dissolution profile with over 95% dissolved by 30 minutes in 0.1 N HCl. This suggests that the differences in bioavailability between the two products were not caused by formulation differences but due to high variability possibly due to highly variable disposition. As exhibited by the AUCT and Cmax data, the inter- and intra-subject cv were well over 30%, indicating propafenone is a highly variable drug. The source of variability can be attributed to the heterogeneity of the subjects (fast vs slow metabolizers) and the presence of saturable first-pass metabolism of the drug in fast metabolizers. The issue of high variability is supported in a preliminary analysis of partial data from a large study in which the reference product was administered to the same subjects on two occasions separated by 1 week. The data indicate that the reference product would have trouble passing all the standard bioequivalence criteria against itself as the inter-period ratios ranged from 0.37-2.09 and 0.39-2.25 for AUCT and Cmax, respectively. To further complicate the picture, propafenone is classified as a drug with a narrower therapeutic range in Canada. This means that the 95% CI is required for both AUCT and Cmax. In addition, a fasting and a food challenge study are needed. The data we have questions the appropriateness of these requirements from an ethical and a cost point of view, as a lot of subjects will be needed. Influence
of Equilibration Diet on the Intra-Subject Variability of a Highly Variable Drug In order to investigate the influence of diet on intra-subject variability of a drug product (Product A), a single-dose, two-treatment, four-period, two-sequence, crossover replicate pharmacokinetic study was conducted using eight healthy volunteers. Control diets (diet 1 and diet 2) were given to the subjects for a total of four days, i.e., from 48 hours before drug administration to 48 hours post drug. Product A was administered twice to each individual while on diet 1 or diet 2. Subjects were crossed over to the alternate diet after the first two periods. The washout between the two drug administrations for each diet (Period 1-Period 2 and Period 3-Period 4) was one week whereas the washout between the two diets (Period2-Period3) was two weeks. Subjects were institutionalized in Period 1 through Period 2 and then Period 3 through Period 4. Blood samples were collected under subdued lighting over 48 h following each drug administration. Drug plasma concentrations were determined by GC-ECD. Faeces samples were also collected up to a maximum of 72 hours post-drug. Analysis of Variance performed on the primary pharmacokinetic parameters detected significant differences between regimens and between replicate arms of each regimen for the dose-dependent parameters relevant to bioequivalence determination, AUC and Cmax. Based on the intra-subject C.V.s, determined from ANOVA residual sum of squares, diet 2 produced less intra-subject differences in the absorption of the drug relative to diet 1. The type of equilibration diet was found to have a profound effect on the intra-subject variability of Product A. Comparison
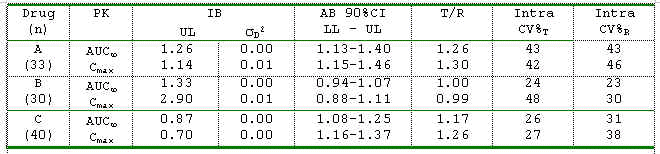
Between Individual and Average Bioequivalence for 3 Highly Variable Drugs The individual bioequivalence (IB) method is now being examined to assess switchability of formulations and as a scientifically improved method for regulatory assessment of bioequivalence. Three fully replicated bioequivalence studies were analysed to compare IB to the current average bioequivalence (AB) method. For IB, the metric suggested by the FDA draft guidance was used along with a 95% Bootstrap upper limit (UL). As depicted in the table, all three drugs have good switchability indicator between test and reference products as the subject*formulation interaction variance component, sD2, is close to 0. Based on the 2.5 limit suggested by the FDA draft guidance, the results indicate that these drugs may meet the regulatory requirements using IB, but may not pass the criteria based on AB, or, vice versa. A test product with an intrasubject CV smaller than the reference product has a better chance to pass IB criterion.
Recommendations for Comparative Bioavailability Standards For
Drugs with a Narrow Therapeutic Range: Experience with Cyclosporine Standards for comparative bioavailability of oral formulations were initiated 25 years ago over concerns about the therapeutic equivalence of apparently similar pharmaceutical products. These standards have evolved from an initial one limited to the requirement of establishing a bioavailability of 80 percent or more of the reference standard to be considered satisfactory. Revised standard for "uncomplicated" drugs were adopted in Canada in 1990. In proposing the new standards the Expert Advisory Committee on Bioavailability indicated that further refinements of these standards should be required for drugs with special characteristics. The Health Protection Branch has circulated in 1997 recommendations for tighter standards for drugs with a narrow therapeutic range. It is evident that many narrow therapeutic drugs have other characteristics either in their pharmacokinetics or in their recommended dose schedules that differentiate them from the so-called "uncomplicated" drugs. Cyclosporine is an example of one such drug. Cyclosporine is a product with complex pharmaceutical characteristics, variable absorption affected by food, and complex kinetics which are dose dependent and time dependent. The drug has a long half life and a complex metabolism with some metabolites being active. Furthermore, the drug is used in conditions which modify its bioavailability. More rigorous standards for comparative bioavailability of cyclosporine are recommended. Individual
Bioequivalence: Attractive Rationale, Practical Difficulties FDA has recently published a Draft Guidance on the possible adoption of individual bioequivalence (IBE). This approach is intended to supersede the procedure of average bioequivalence (ABE) which is presently applied. The rationale for the suggestion of IBE is interesting. First, the switching of formulations within individuals is considered instead of prescribing drugs based on the comparison of average kinetic measures. IBE contrasts not only these averages but evaluates also the subject-by-formulation interaction as well as the intrasubject variations of the two contrasted formulations. The models combining these terms (components) are expected to satisfy either an unscaled or, for highly variable drugs, a reference-scaled regulatory criterion. To date, only few investigations have explored properties of the suggested IBE models. However, results of all of these studies strongly question whether the IBE models yield practically reasonable conclusions. Because of the interplay (tradeoff) among the components of the models, the deviation between the two averages which is still compatible with the declaration of bioequivalence, can be very much expanded or contracted just by random chance. The reference-scaled model admits bioequivalence liberally and is very insensitive to the difference between the averages. Consequently, the practical usefulness of the proposed models for IBE is questioned. The possible adoption of the proposed approach should not be considered at least until the completion of extensive further investigations. Sample
Size Considerations for Bioequivalence Studies of Narrow Therapeutic Range Drugs Acceptance criteria for bioequivalence of narrow therapeutic range (NTR) drugs, as recommended by various regulatory agencies, are based on a choice between two approaches. First is to adopt a tighter acceptance range (e.g., 85-118% or 90-111%) than 80-125% without changing the confidence level (90%), and second is to use a 95% confidence interval (CI) instead of a 90% CI for relative mean AUC and Cmax. These two approaches have different rationales, different chances of product acceptability, and different sample size (number of subjects) requirements. We determined the number of subjects required in a BE study to meet a given standard for both approaches. All calculations were done for a wide range (15-35%) of intrasubject co-efficient of variation (CV) and up to 10% difference in the test-to-reference ratio of means, to achieve a power of 80% to demonstrate bioequivalence. For 2-period, 2-treatment studies of NTR drugs with intrasubject CV up to 35%, sample size requirements are not very different for meeting 90% or 95% CI standards. However, for meeting a tighter range of acceptance, substantially higher number of subjects is needed compared to the current requirements of 90% CI being within 80-125% range. For example, relative to an acceptance range of 80-125%, 3 times as many subjects will be required to pass the 85-118% range for a 10% difference in the test-to-reference ratio of means. Our calculations also suggest that passing the 90-111% acceptance range for a 10% difference in the test-to-reference ratio of means will not be practically possible since too high a number of subjects will be required. Narrow
Therapeutic Range Drugs: Experiences with Clozapine Clozaril (clozapine) is indicated for the management of symptoms of treatment-resistant schizophrenia. Clozapine is classified as an atypical antipsychotic drug because its profile of binding to dopamine receptors and its effects on various dopamine mediated behaviours differ from those exhibited by more typical antipsychotic drug products. Like other tricyclic antipsychotics, clozapine shares some of their side effects. However, Clozaril has some serious side effects that must be monitored during treatment. Specific warnings are given to the occurrence of agranulocytosis, seizures and orthostatic hypotension with and without syncope. Thus, clozapine can be considered a drug with a narrow therapeutic range. Through more than 30 years experience with clozapine, Novartis has gained extensive knowledge and experience concerning the safety and efficacy of clozapine. This knowledge and experience included the results of many clinical and bioequivalence studies, pre- and post-market patient monitoring and other clinical observations. Serious risks of cardiac arrest and other cardiovascular problems with healthy subjects involved in clozapine bioequivalent studies have been reported. Similar adverse events have also been reported with mild to moderate schizophrenic patients. However, the reported adverse effects were not as severe as those reported in the healthy subjects. As a result, Novartis has come to the conclusion that in order to avoid undue life-threatening adverse effects in healthy subjects, all bioequivalent and pharmacokinetic studies should be conducted in treatment resistant schizophrenic patients. The Importance
of Tight Manufacturing Specifications in Narrow Therapeutic Range Drugs This presentation will look at the importance of tight manufacturing specifications in warfarin sodium tablets, a narrow therapeutic range (NTR) drug, and dosage form uniformity as a critical component of a product specification. Specifications are set forth in the USP for determination of dosage form uniformity by assay of individual units for the active ingredient(s) in a single tablet. Current USP requirements allow for a + 15% variability in table potency. In the case of the wider specification of warfarin sodium tablets with nine strengths, this may cause an overlap of drug potency in various table strengths. A review of additional stringent testing to ensure dose, intra-batch and batch to batch uniformity in the formulation of warfarin sodium tablets along with historical information of batches produced will also be presented. Is Fuzzy
Logic too Fuzzy for Bioequivalence Assessment? It is common for humans to recognize and reason through vague assertions or claims. To hear imprecise remarks like "it will be cloudy", "the conference will be illuminating", or "the creatinine clearance is normal" brings us face to face with fuzzy terminology. It is contrasted by so-called "crisp" distinctions that are precise and unambiguous; example: the temperature of the oven is 365 degrees Fahrenheit. Ordinarily, we mentally manipulate fuzzy statements and arrive at an interpretation that is seemingly understood. The understanding may however not be the same for all. Fuzzy logic modelling employs a methodology, strongly influenced by IF-THEN rules (or commonsense rules), to arrive at a reasoned understanding when fuzzy datasets are encountered. This approach has been employed rather successfully in the engineering field such that the use of machine intelligence has enabled computers to arrive at the desirable outcome. For example, in recent years energy efficient refrigerators have built in which the input and output devices employ such an approach. In bioequivalence, it is evident that fuzzy datasets have arisen and fuzzy terminology has been adopted. This encompasses such concepts like "bioequivalent", "narrow therapeutic index", "highly variable", "nonlinear", "prescribable", and "switchable". The current tension between average bioequivalence and individual bioequivalence illustrates the dilemma of decision-making wherein the understanding is different for those who specialize in statistics, pharmacokinetics, or clinical medicine. Furthermore, it is evident that some terms in this field need to be reexamined and defuzzified. In view of the fact that bioequivalence entails data sets that are not crisp, we propose that a form of fuzzy logic needs to be adopted. This will help to create a reasoned approach to bioequivalence that will permit regulatory bodies to set appropriate standards for drugs (new and perhaps old) and assist competitive manufacturers in developing a reasoned approach in product development. Bioequivalence of Chiral Drugs: an Update Many commonly used drugs are chiral and available as racemate. Enantiomers of racemic drugs, however, may differ from each other in terms of pharmacodynamic and/or pharmacokinetic properties. When the enantiomers are equipotent, the issue of stereoselectivity in drug development becomes less important unless under certain pathophysiological conditions when clinically significant stereospecific or stereoselective altered pharmacokinetics are observed. Similarly when enantiomers posses different pharmacodynamics but little or no pharmacokinetic differences, non-sterospecific monitoring of the drug reflects the time-course of either individual enantiomers. On the other hand, stereochemistry is of importance when bioequivalence is assessed for following products:
In the past decade many regulatory guidelines have been presented which deal with the issue of stereochemical aspects of drug development. Interestingly, however, very few bioequivalence assessments have been made using stereospecific approaches. This is mainly due to the fact that the pharmacokinetic evaluations of the innovator product are usually carried out using non-stereospecific assays. Indeed, at the time of regulatory review of many of the available racemic products, the issue of stereochemistry in pharmacokinetics was only a topic of future. Stereochemistry in bioequivalence, however, will progressively become a more important regulatory issue since presently many new drug applications include pharmacokinetic data based on stereospecific assays. It is, therefore, expected that subsequent bioequivalence assessment to be made using approaches similar to those of the innovator products. Recent studies have indicated that the presence of an enantiomer, however with no intrinsic effect, may influence the therapeutic outcome of the pharmacologically ‘active’ enantiomer. This interaction may be of physicochemical, pharmacokinetic and/or pharmacodynamic nature. Hence a stereochemically pure "active" enantiomer may not be a mere more refined form of the racemate. In assessing bioequivalence of chiral drugs, therefore, plasma concentration of all enantiomers should be evaluated regardless of their relative potency. Bioequivalence of Non-Linear Drugs: Saturation of
Plasma Proteins A draft Drugs Programme Policy on bioequivalency of drugs having non-linear kinetics was circulated by the HPB early last year. Two categories of nonlinear drugs were identified: those showing greater than dose proportional increases in AUC, and those showing less than dose proportional increases in AUC. It was recommended that bioavailability comparisons concentrate on the highest strength of the product when greater than proportional increases are observed, and on the lowest strength of the product when less than proportional increases are observed. The rationale being that under those test conditions, inequalities in bioavailability will maximized in the AUC determinations. However, the mechanism(s) responsible for a non-linearity should also influence the decision. Drugs that saturate their available receptors at standard doses do not necessarily show their greatest sensitivity to bioavailability differences under the conditions suggested. Nonlinear
Kinetics and Pharmacological Response The influence of zero-order kinetics of a drug on its pharmacological response depends upon the cause of the nonlinearity. Zero-order absorption does not affect the pharmacokinetic-pharmacodynamic relationship (PK/PD). Changes in the drug’s volume of distribution due to plasma or tissue binding modifications may affect PK/PD. Finally, PK/PD will be affected by changes in the clearance whenever the activity of the metabolites is greater than that of the parent compound. To test the affect of zero-order elimination on the response of a drug that generate multiple active metabolites, we used mibefradil, a calcium channel blocker. An open parallel study was conducted with two groups of 10 healthy volunteers. On days 1 and 8, 10 volunteers were given 50 mg p.o. and 5 mg of deuterated mibefradil through a 10 minutes i.v. infusion. On days 3 to 7, they received 50 mg of mibefradil orally daily. The remaining 10 volunteers observed the same protocol, except that the oral doses of mibefradil were 100 mg. Serial blood samples were withdrawn for 48 hours, and mibefradil plasma concentrations were assayed by LC-MS. Blood pressure and heart rate were measured for 4 hours, and a 12 leads ECG was performed 2 hours after drug administration. Repeated oral administrations of 50 mg of mibefradil to steady state generated zero-order kinetics secondary to a decrease in mibefradil systemic clearance. Compared to the 50 mg dose, a single oral dose of 100 mg decreased the systemic clearance of mibefradil, and repeated oral administrations of 100 mg of mibefradil to steady state also generated zero-order kinetics leading to a further decrease in mibefradil systemic clearance. The oral bioavailability of mibefradil was not affected by the presence of zero-order kinetics. The 50 mg dose of mibefradil did not affect blood pressure or the ECG. Mean diastolic blood pressure was decreased by the single 100 mg dose, response that was not increased by a two-fold increase in mibefradil plasma concentrations at steady state. Heart rate was decreased at day 8 by the 100 mg dose. Compared to the 50 mg dose, the QT interval was increased by multiple 100 mg doses. These results suggest that despite zero-order kinetics, the pharmacological response to mibefradil can be predicted, i.e. the response increases proportionally to the dose.
Published by the Canadian Society for Pharmaceutical Sciences. Copyright © 1997 by the Canadian Society for Pharmaceutical Sciences. |