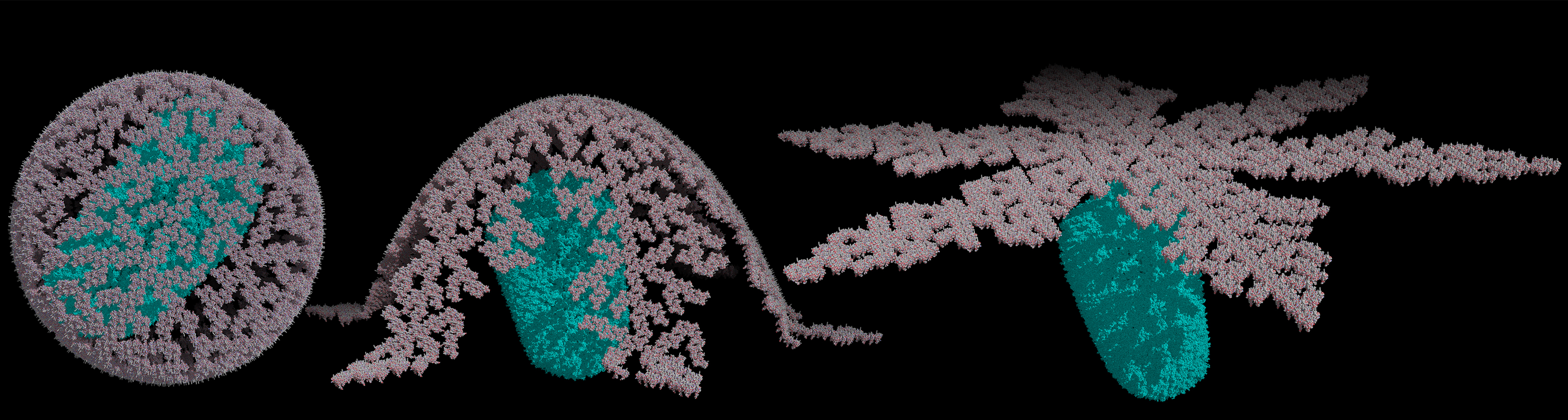
This site will provide you with the tools and step-by-step guide to build HIV-1 Matrix (MA) shells using methods described by Sun W, et al. (2019) Mathematical determination of the HIV-1 matrix shell structure and its impact on the biology of HIV-1. PLOS ONE 14(11): e0224965.
We will introduce you to 4 scripts, each of which will allow you to develop unique outcomes:
Calculate trimer coordinations. Here the position of the center-of-mass for each trimer is placed on a sphere.
Generate PyMol Model using the output file from coordinates above.
Generate PyMol Flatten Model using coordinates above.
Generate tabulated relationship between trimer numbers per viral particle and radius of HIV-1 MA shell.
Python 3.5 or greater
Python Env Library in Jupytor NoteBook:
Basemap 1.2.0 or greater
PyMol 2.3.2 or greater
Pandas 0.24.2 or greater
Open Anaconda and create a python 3.5 environment
Install basemap, PyMol, pandas libraries
If a library(s) is not in your Anaconda package, follow the commands bellow
conda install -c anaconda basemap
conda install -c anaconda pandas
conda install -c schrodinger pymolThen open the main_flow.ipynb file.
You should run the code successfully as shown here.
Note: We recommend using the Anaconda environment system described under "Getting Started". The instructions under "Python 2.7 Support" do not need to be followed if you used the Anaconda environment described above.
Check prerequisites
Python 2.7
Python Libraries: importlib, matplotlib, mpl_toolkits
>python calculate_trimer_coordinates.py
Input the R:
632
Do you want to use default r = 52.0769942809? (y or n):
yThen you will receive the following message and two output files >> 'points' and 'lines'
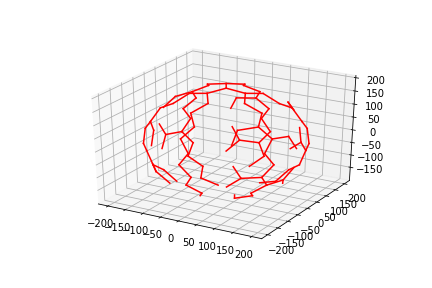
There are 1024 points.There are 1024 points' coordinations in the points file and there is another file lines that provides the relationship between the center-of-mass of each trimer and their coordination.
You will be given an option to quickly view what the plots look like. You could input 3 to quit this script.
1 - show and save the lines plot
2 - show and save the points model
3 - quit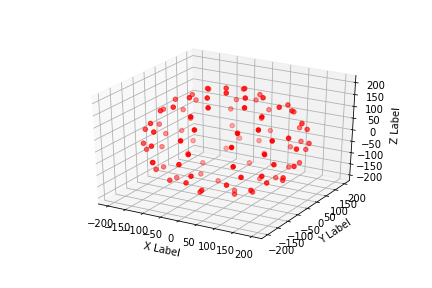
python pymol_plot_trimer.pyIf your machine supports PyMol, then PyMol will open and run the script within pymol itself.
After the models are generated in PyMol, you may store your models in pdb or pse file formats from PyMol.
> python calculate_flatten_trimmer_coordinations.pyWhich will transfer points output file to points-2D
Then you run
> python pymol_2D_plot_trimer.pyAfter the models are generated in PyMol, you may store your models in pdb or pse file formats from PyMol.
> python tabulate_relationship_of_R_and_trimer_number.pyThen you need to input minimum (Min) Radius, maximum (Max) Radius, and size of the interval steps (interval). The default r referse to the distance between adjacent trimers. All units are in Angstroms.
E.g.:
Input the Min Radius: 600
Input the Max Radius: 700
Input the interval: 10
Do you want to use default r = 52.0769942809? (y or n): yThe script will output how many trimer units can fit in spheres for each HIV-1 MA radius.
E.g.:
909
955
971
1019
1033
1074
1124
1157
1178
1185
1243ACADEMIC PUBLIC LICENSE: This license contains the terms and conditions of using HIV-1 Matrix Builder in noncommercial settings: at academic institutions for teaching and research use, and at non-profit research organizations. You will find that this license provides noncommercial users of HIV-1 Matrix Builder with rights that are similar to the well-known GNU General Public License (GPL), yet it retains the possibility for HIV-1 Matrix Builder authors to financially support the development by selling commercial licenses. In fact, if you intend to use HIV-1 Matrix Builder in a "for-profit" environment, where research is conducted to develop or enhance a product, is used in a commercial service offering, or when a commercial company uses HIV-1 Matrix Builder to participate in a research project (for example government-funded or EU-funded research projects), then you need to obtain a commercial license for HIV-1 Matrix Builder. In that case, please contact the Author to inquire about commercial licenses.
What are the rights given to noncommercial users? Similarly to GPL, you have the right to use the software, to distribute copies, to receive source code, to change the software and distribute your modifications or the modified software. Also similarly to the GPL, if you distribute verbatim or modified copies of this software, they must be distributed under this license.
By modeling the GPL, this license guarantees that you're safe when using HIV-1 Matrix Builder in your work, for teaching or research. This license guarantees that HIV-1 Matrix Builder will remain available free of charge for nonprofit use. You can modify HIV-1 Matrix Builder to your purposes, and you can also share your modifications. Even in the unlikely case of the authors abandoning HIV-1 Matrix Builder entirely, this license permits anyone to continue developing it from the last release, and to create further releases under this license.
Weijie Sun, Eduardo Reyes-Serratos, David Barilla, Joy Ramielle L. Santos, Mattéa Bujold, Sean Graves and Marcelo Marcet-Palacios. (2019) Mathematical determination of the HIV-1 matrix shell structure and its impact on the biology of HIV-1. PLOS ONE 14(11): e0224965.
Please contact Weijie Sun at weijie2@ualberta.ca for technical support.
Please contact Dr. Marcelo Marcet-Palacios at marcelo@ualberta.ca for commercial license inquiries.