J Pharm Pharmaceut Sci (www.cspscanada.org) 9(2):140-148, 2006
Formulation modifications of PD 0313052, a direct Factor Xa Inhibitor, alter pharmacokinetics and pharma-codynamics following subcutaneous administration to rabbits.
Yun-Wen Peng, Liguo Chi, Glenn Gibson, Nancy Janiczek, Paul Juneau, Daniel Ross, Lisa A. Perrin, and Robert Leadley
Pfizer Global R&D, Michigan Laboratories, Ann Arbor, Michigan, USA
Cardiovascular Biology, Pharmacokinetics Dynamics and Metabolism, Biostatistics, Antibacterial Biology, Pfizer Global R&D, Michigan Laboratories, 2800 Plymouth Road, Ann Arbor, Michigan, USA
Date received January 27, 2006, revised version received April 27,2006; accepted April 30, 2006; published May 5, 2006.
Corresponding Author: Robert Leadley, Ph.D, Pfizer Global R&D, Michigan Laboratories Cardiovascular Biology 2800 Plymouth Road Ann Arbor, Michigan 48105, USA Phone: (734) 622-1420 FAX: (734) 622-1480 E-mail: robert.leadley@pfizer.com
ABSTRACT: PURPOSE PD 0313052 is a potent, direct factor Xa (FXa) inhibitor (Ki = 0.33 nM) and its antithrombotic effect has been previously demonstrated in several animal models, via intravenous (IV) administration. In the present study, we evaluated four different subcutaneous (SC) formulations to test the feasibility of developing PD 0313052 as a subcutaneous agent. METHODS: PD 0313052 was formulated in saline, methylcellulose (MC, 0.5% methylcellulose solution containing 1% Tween-80), sesame oil, and F127 (25% aqueous solution). Each formulation was injected subcutaneously into rabbits and the relative plasma exposure and the duration of action of PD 0313052 were assessed. Plasma concentration, FXa activity, and coagulation parameters were used to monitor the pharmacokinetic (PK) and pharmacodynamic (PD) profiles of PD 0313052. RESULTS: Regardless of formulation, there was a significant (p<0.05) correlation between PD 0313052 plasma concentration and FXa activity (R2 = 0.90), prothrombin time (PT) (R2 = 0.86), and Heptest (R2 = 0.93). The saline and MC formulations had similar effects on FXa activity, coagulation parameters, and Heptest, peaking at 30 to 120 minutes after administration and decreasing rapidly thereafter. In contrast, formulations of F127 and sesame oil yielded lower maximal effects on PD markers but produced sustained PD effects over time. CONCLUSION: The data indicate that PD 0313052 is bioavailable after SC administration to rabbits and that there is a strong correlation between the PD parameters and plasma concentrations of PD 0313052. Modifications in the formulation of PD 0313052 produce marked differences in the PK and PD profiles of this agent after SC administration to rabbits. These results suggest that SC formulations can be optimized to improve the PK and PD profiles of PD 0313052, and that PD 0313052 is a viable candidate for development as a SC antithrombotic agent.
Introduction
Although heparin and low-molecular-weight heparin are long-established treatments for thrombotic diseases, they indirectly (via anti-thrombin III) target the coagulation cascade at multiple sites and have pharmacokinetic (PK) and pharmacodynamic (PD) relationships that are complex and variable. Frequent laboratory monitoring is often required and dosage requirements for heparin vary from patient to patient. In addition, side effects, limitations of efficacy, and bleeding complications with these agents have stimulated a long and intense search for more direct and specific inhibitors of key enzymes in the coagulation cascade [1-4]. For example, selective inhibition of factor Xa (FXa) has become an attractive target for developing antithrombotic therapy because of its central and upstream position in the coagulation process [5]. FXa plays a central role in the coagulation cascade, linking the extrinsic and intrinsic pathways by catalyzing the conversion of prothrombin to thrombin on the cell surface.
FXa inhibitors with direct enzyme inhibitory action, more predictable PK and PD profiles, fixed and simpler dosing regimens, and few or no laboratory monitoring requirements would be ideal replacements for heparins. A number of small molecule, direct FXa inhibitors effectively inhibit thrombus formation in animal models [6-8] and some have entered clinical trials [9, 10]. To develop an orally active direct FXa inhibitor is desirable; however, success in creating an acceptable orally active FXa inhibitor has been limited due to the requirement for benzamidine or benzamidine derivatives to achieve adequate potency of these agents. The benzamidine moiety is commonly associated with poor oral bioavailability and a short duration of action [11-13]. Clearly, orally-active FXa inhibitors would be a major breakthrough in antithrombotic therapy; however, the ability to deliver potent, selective FXa inhibitors via subcutaneous administration would also be beneficial. For example, subcutaneous (SC) administration may simplify patient treatment in or out of the hospital and would be particularly advantageous for patients who could not be treated easily by oral or IV administration (e.g. heart attack, stroke, or unconscious patients).
PD 0313052 (Figure 1) is a potent (Ki = 0.33 nM), selective (10,000‑fold selective versus other serine proteases), and direct inhibitor of human FXa [14]. PD 0313052 effectively inhibited thrombus formation in a canine model of arterial thrombosis after intravenous administration [15]. In the present study, we investigated the feasibility of subcutaneous administration of PD 0313052 in a rabbit model and compared four common pharmaceutical formulations. The data indicate a strong correlation between the PD parameters and plasma concentration of PD 0313052. These studies demonstrate that PD 0313052 can be effectively administered subcutaneously in the rabbit and that different formulations of PD 0313052 can produce significant alterations in the PK and PD profiles.
MATERIALS AND METHODS
Materials
PD 0313052,2‑(5‑carbamimidoyl-2‑hydroxy-phenyl)-4‑[5‑(2,6‑dimethyl-piperidin-1‑yl)-pentyl]-3‑oxo-3, 4‑dihydro-quinoxaline-6‑carboxylic acid, was synthesized by the Department of Chemistry, Pfizer Global R&D, Ann Arbor Laboratories.

Figure 1. The chemical structure of PD 0313052: 2‑(5‑carbamimidoyl-2‑hydroxy-phenyl) 4‑[5‑(2,6‑dimethyl-piperidin-1‑yl)-pentyl]-3‑oxo-3, 4‑dihydro-quinoxaline-6‑carboxylic acid.
Formulations
Saline (Baxter, Deerfield, IL) served as an immediate-release water-soluble formulation and sesame oil (Sigma, St. Louis, MO) is a delayed-release oil suspension [16]. The methylcellulose (MC) formulation was prepared from 0.5% MC (Sigma, St. Louis, MO) mixed with 1% Tween-80 (Sigma, St. Louis, MO). An aqueous solution of F127 (25%, BASF, Mount Olive, NJ), a propylene oxide copolymer, was used for the formulation of emulsion for drug release [17, 18]. PD 0313052 was also administered intravenously in saline.
Animals
The in vivo experiments were conducted in accordance with the Institutional Animal Care and Use Committee of Pfizer Global Research and Development, Ann Arbor Laboratories, according to the NIH Guidelines for the Care and Use of Laboratory Animals. The rabbit was selected as the model for these studies because rabbit and human FXa have similar binding affinities to enzyme substrate and to small molecule inhibitors of FXa [19]. Male New Zealand white rabbits (2.5‑3.2 kg) were anesthetized by IV injection of sodium pentobarbital (30 mg/kg) through an ear vein catheter. Anesthesia was maintained with intermittent administration of sodium pentobarbital intravenously during the experimental procedure.
The treatment groups or vehicle (saline, n=4) control were studied in anesthetized rabbits by SC-bolus injection through the abdominal skin using a 22-gauge needle. The four treatment groups were as follows: PD 0313052 (3 mg/kg) dissolved in saline (n = 4), MC (n = 4), sesame oil (n = 3), and F127 (n = 3), respectively. As a reference control, PD 0313052 was administered i.v. to another group of animals (n=5). Blood samples were collected at baseline, 10, 15, 30, 60, 90, 120, 150, 180, 240, 300, and 360 minutes after SC administration. Bleeding times were measured at baseline and 60 minutes. For the i.v. experiments, blood samples were obtained at 5, 10, 15, 30, 60, 90, and 120 minutes after dosing and bleeding times were determined at 5 and 120 minutes after dosing. The ex vivo coagulation parameters (PT, aPTT, and ACT), FXa activity, and Heptest were measured using the methods described below.
Ex vivo Coagulation Assays and Bleeding Times
For determination of coagulation status, blood samples (1.8 mL) were drawn into a syringe containing 0.2 mL of 3.8% sodium citrate (1:10 dilution) then centrifuged at 2,000 ´ g for 10 minutes to obtain plasma that was used in the assays. PT and aPTT were determined using a MCA210‑Micro Coagulation Analyzer (Bio/Data, Horsham, PA) with the reagents Innovin and Actin FS (Dade Behring, Deerfield, IL), respectively. The activated clotting time (ACT) was determined on an ACT II Automated Coagulation Timer II (Medtronic, Inc, Parker, CO) using fresh whole blood and low-range ACT cartridges (Medtronic, Inc, Parker, CO). The reported results are the average of duplicate measurements. To evaluate the risk of bleeding, an ear bleeding time technique was adapted from Hollenbach, et al. [20]. Briefly, a No. 11 scalpel blade was inserted through the ear between the central ear artery and the marginal ear vein. Blood was blotted from the wound with Whatman #2 filter paper (Whatman International Ltd, England) every 10 seconds until no blood was transferred to the filter paper. Bleeding time was determined from the moment of the incision until the blood no longer stained the filter paper.
Ex vivo FXa Activity Assay, Heptest, and Plasma Concentration of PD 0313052
The FXa activity assay was performed utilizing Actichrome Heparin (American Diagnostic, Greenwich, CT) following the manufacturer’s instructions. The assay measures proteolytic activity of enzyme by cleavage of a paranitroanilide (pNA) substrate in a 96‑well microtiter plate. The rate of change in absorbance at 405 nm was monitored by a Vmax Microplate Reader (Molecular Devices, Sunnyvale, CA) to determine initial rates of substrate hydrolysis (DA405/min). These initial rates were converted to percent inhibition of FXa activity by comparison to a baseline sample. The Heptest is a clotting assay which measures FXa activity indirectly. The principle of the Heptest (American Diagnostica, Greenwich, CT) is the ability of a test agent to inhibit exogenous bovine factor Xa in the presence of CaCl2 and brain cephalin. The extent of FXa inhibition is directly proportional to the prolongation of the clotting time of the plasma sample [21, 22]. The time for clot formation was measured using an automatic coagulometer (ST4, Diagnostica Stago, Parsippany, NJ). The reported results are the average of duplicate measurements. Plasma concentrations were measured in heparinized plasma samples by LC/MS/MS. The lower limit of quantitation was 10 ng/mL.
Statisitcal Analysis
All data were summarized as the mean ± standard error. The level of statistical significance for all tests was p<0.05. The responses of all formulations were compared to vehicle control in a pair-wise fashion via Dunn’s test [23].
RESULTS
Effects of Different PD 0313052 Formulations on FXa Activity and Heptest
To examine the anti-FXa effect of PD 0313052 in different formulations, PD 0313052 was administered SC at 3 mg/kg and FXa activity was evaluated at several time points. Figure 2A indicates an inhibitory effect on FXa in all formulations evaluated. PD 0313052 formulations of saline and MC had similar time-course profiles. FXa activity peaked at 30 to 120 minutes (approximately 95% inhibition), then decreased to approximately 50 to 60% at 6 hours (p<0.05). In contrast, formulations of sesame oil and F127 showed lower maximal effects on FXa activity. The peak level of inhibition with sesame oil and F127 was 80 to 90%, but the anti-FXa effect of PD 0313052 remained constant over 6 hours. In comparison, i.v. administration of PD 0313052 maximally inhibited FXa activity (98±1%) at 5 minutes after dosing, returning to 41±12% inhibition within 2 hours.
The effect of altering PD 0313052 formulations on the Heptest was also evaluated (Figure 2B). The saline formulation had the highest increase in Heptest, which was 10‑fold over baseline at 30 minutes (p<0.05) and 2‑fold over baseline at 6 hours. In the MC formulation, the maximal change was 8‑fold over baseline at 60 minutes (p<0.05) and was 3‑fold at 6 hours. Compared to formulations of saline and MC, sesame oil and F127 formulations had lower peak effects on the Heptest. The peak change was 3‑ to 4‑fold over baseline at 15 and 360 minutes and was maintained over 6 hours in formulations of sesame oil and F127, respectively. Intravenous administration of PD 0313052 increased the Heptest to 18±2.3-fold over baseline at 5 minutes after dosing, returning to 1.6±0.2-fold at 2 hours.
Effects of Different PD 0313052 Formulations on Coagulation Parameters and Bleeding Time
Prothrombin Time (PT) is a plasma clotting-time assay that can be used to monitor the potency of FXa inhibitors. Data describing the effect of PD 0313052 SC administration on PT are shown in Figure 3A. The formulations of saline and MC caused a similar PT prolongation; PT increased maximally to 4‑fold and 3‑fold over baseline with the formulations of saline and MC, respectively (p<0.05), then gradually returned to baseline at 6 hours. In contrast, the maximum PT prolongation was only 1.6‑fold over baseline with formulations utilizing F127 and sesame oil, but with F127, the PT prolongation was sustained for 6 hours after SC administration. PT increased 8±0.8-fold at 5 minutes after i.v. dosing, returning to 1.3±0.1-fold at 90 minutes
As seen with PT, different formulations of PD 0313052 prolonged aPTT and ACT, but the sensitivity of aPTT and ACT was less than that of PT (Figure 3B and 3C). Formulations of saline and MC increased aPTT and ACT to 2 to 3‑fold of baseline at 1 hour after administration (p<0.05) and gradually returned toward baseline over 6 hours. In contrast, formulations of F127 and sesame oil produced maximinal aPTT and ACT prolongations of 1.5‑fold over baseline, which were sustained for 6 hours after SC administration. Maximal increases in aPTT and ACT (4.4±0.6 and 4.5±0.5-fold, respectively) were observed 5 minutes after i.v. administration and returned to near baseline at 2 hr after dosing.
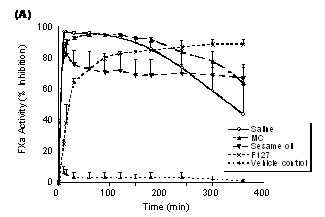
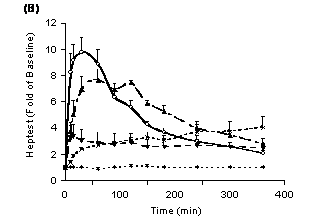
Figure 2. Different formulations of PD 0313052 (3 mg/kg) were subcutaneously administered to rabbits (Mean + SEM, n=3 or 4). A: Plasma FXa activity was evaluated at multiple time points. Saline and MC peaked at 30 to 120 minutes, then decreased to approximately 50 to 60% at 6 hours (p<0.05). Sesame oil and F127 showed lower maximal effects on anti-FXa activity, but the effect remained constant over 6 hours. B: Heptest was evaluated at multiple time points. Saline formulation yielded a 10‑fold increase in Heptest over baseline at 30 minutes (p<0.05) and MC increased the Heptest 8‑fold over baseline at 60 minutes (p<0.05). Compared to saline and MC, sesame oil and F127 had lower peak effects on the Heptest.
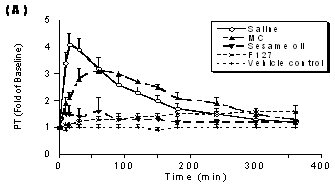
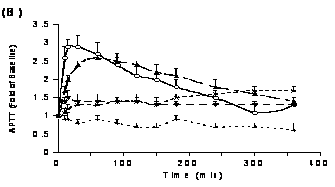
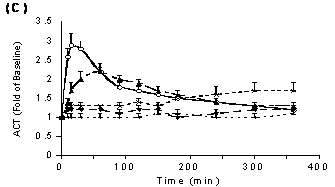
Figure 3. The effects of different formulations of PD 0313052 (3 mg/kg) on coagulation parameters (Mean + SEM, n=3 or 4) were determined in plasma samples that were collected at multiple time points. A: PT profiles. Saline and MC formulations increased PT maximally to 4‑fold and 3‑fold over baseline, respectively (p<0.05). The maximum PT prolongation was only 1.6‑fold over baseline with F127 and sesame oil. B: APTT profiles. Saline and MC formulations yielded aPTTs 2 to 3‑fold over baseline at 1 hour (p<0.05). F127 and sesame oil produced maximinal aPTT prolongations of 1.5‑fold over baseline. C: ACT profiles. Like APTT, saline and MC formulations resulted in ACT increases of 2 to 3‑fold over baseline at 1 hour (p<0.05). Formulations of F127 and sesame oil yielded modest effects on ACT.
The bleeding time was significantly increased 4‑ to 5‑fold over the baseline at the first hour and was 2‑ to 2.5‑fold over baseline at 6 hours with the formulations of MC and saline, respectively (data not shown). In formulations of sesame oil and F127, bleeding times did not increase significantly from pre-drug values. Intravenous administration resulted in a 4.8±3.1-fold increase in bleeding time at 5 minutes and 2.6±0.2-fold at 2 hours after dosing.
Effects of Different Formulations on PD 0313052 Plasma Concentrations
The plasma concentrations of PD 0313052 resulting from administering different formulations were determined by LC/MS/MS (Figure 4). Considering s.c. formulations, saline yielded the highest peak plasma concentration at 30 minutes (7,000 ng/mL). Compared with the saline formulation, the peak plasma concentrations were lower in formulations of MC (2,600 ng/mL at 1 hour), F127 (700 ng/mL at 6 hours) and sesame oil (600 ng/mL at 0.25 hours). However, formulations of F127 still maintained plasma concentrations of 700 ng/mL at 6 hours after dosing. The Cmax for i.v. administration was 17,630±3119 ng/mL at 5 minutes, decreasing to 144±27 ng/mL at 2 hours.
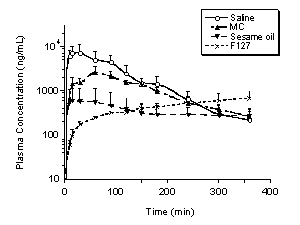
Figure 4. Plasma concentrations of PD 0313052 (ng/mL) following subcutaneous administration of PD 0313052 (3 mg/kg) in rabbits were evaluated at multiple time points (Mean + SEM, n=3 or 4). Saline had the highest peak plasma concentration at 30 minutes (7000 ng/mL). The peak plasma concentrations were lower in MC (2600 ng/mL at 1 hour), F127 (700 ng/mL at 6 hours) and sesame oil (600 ng/mL at 0.25 hours).
Correlation of PD 0313052 Plasma Concentrations with FXa Activity, Prothrombin Time, and Heptest
There was a significant (p<0.05) correlation between PD 0313052 plasma concentration and FXa activity, PT and Heptest with all formulations. PD 0313052 produced concentration-dependent ex vivo FXa inhibition with an IC50 of 164 ng/mL (R2 = 0.90). As shown in Figure 5, PD 0313052 plasma levels also correlated well with the PT fold-change (R2 = 0.86) and with the Heptest fold-change (R2 =0.93). These results suggest that FXa activity, PT, and Heptest can be used as reliable PD markers for PD 0313052.
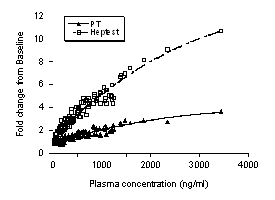
Figure 5. Correlation between PD markers (PT and Heptest) and plasma PD 0313052 concentrations in all formulations. The R2 values are 0.86 and 0.93 for PT and Heptest, respectively (p<0.05).
DISCUSSION
To evaluate the feasibility of developing PD 0313052 as a SC agent, four different types of pharmaceutical formulations of PD 0313052 were evaluated including an immediate-release water-soluble formulation (saline), a polymeric gel (F127), an emulsion (MC), and an oil suspension (sesame oil). Plasma concentration and multiple PD markers (FXa activity, coagulation parameters, and Heptest) were investigated in the same compartment (plasma). The data showed that the plasma concentration of PD 0313052 significantly correlated with FXa activity, PT, and Heptest, regardless of the formulation. The results confirm that SC formulations of PD 0313052 can provide favorable PK and PD profiles in the rabbits. Formulations of saline and MC produced the most pronounced, albeit short-lived, effects on FXa activity, coagulation parameters, and Heptest. In contrast, formulations of sesame oil and F127 exhibited lower peak effects on PK and PD markers, but produced more sustained effects over time. Due to the potential for bleeding complications with a FXa inhibitor, a small peak-to- trough ratio of plasma concentration is desirable. The current results suggest that formulations similar to sesame oil or F127 would be well suited for reducing PK/PD variability, thereby avoiding potentially dangerous high drug levels as observed with i.v. administration of PD 0313052. Optimization of these, or other, formulations would likely yield a delivery option that would prolong the duration of action of PD 0313052 without producing excessive peak drug concentrations. Consequently, SC formulations of PD 0313052 are not only feasible, but may produce a safe and effective delivery system for this antithrombotic agent.
It is important that PK/PD relationships for multiple coagulation markers be investigated during drug development. The correlation between PK and PD relate the drug’s exposure in subjects with the agent’s anticoagulant effect (safety) and anti-thrombotic outcome (efficacy). Synthetic, selective, direct inhibitors of clotting factors, such as thrombin or FXa are attractive antithrombotic approaches because of favorable and predictable PK and PD profiles. This study indicated a close correlation between PD 0313052 plasma concentration and PD markers (R2 = 0.89, 0.86 and 0.93 for FXa activity, PT and Heptest, respectively). The results of this study suggest that FXa activity, PT and Heptest could be used to characterize the anticoagulant effect of PD 0313052. These observations of PK and PD are predictable and the characteristics are similar to other small molecule, direct FXa inhibitors such as CI-1031 (ZK-807834) and BAY 59-7939 in animal models of thrombosis in rats, dogs and rabbits [24, 25].
Formulation development and optimization is a key factor for effective subcutaneous drug delivery. The formulation chosen must be able to solubilize the drug at the desired concentration and must provide an environment where the drug has sufficient chemical stability. SC administration requires a low viscosity formulation, a reduced injection volume to less than 2 ml, and restrictions on both the pH range and the amount of solvent due to slower diffusion away from the injection site [26]. In this study, four types of formulations of PD 0313052 were investigated in rabbits. For all formulations, PD 0313052 plasma concentration and FXa activity, PT, and Heptest indicate a significant correlation. The results suggest that PD 0313052 can be effectively formulated for SC administration.
Recently, several FXa inhibitors have been designed to provide safe and effective therapy for prevention and treatment of venous and arterial thromboembolism [27-30]. These novel FXa inhibitors include the subcutaneous indirect FXa inhibitors fondaparinux and idraparinux and the orally active direct FXa inhibitors razaxaban, BAY 59-7939, and LY 517717. Of these agents, only fondaparinux is currently available in the United States and the orally active agents are in various stages of clinical development [31-33]. The feasibility of formulating these oral agents for subcutaneous administration is uncertain. Consequently, a subcutaneous formulation of potent and selective FXa inhibitors such as PD313052 would fulfill a currently unmet medical need for immediate and simple antithrombotic therapy.
Conclusions
To evaluate the feasibility of developing PD 0313052 as a SC agent, four different formulations of PD 0313052 were evaluated in a rabbit model. Plasma concentration, FXa activity, and coagulation parameters were used to monitor the pharmacokinetic (PK) and pharmacodynamic (PD) profiles of PD 0313052. The data indicate a significant correlation between PD 0313052 plasma concentration and FXa activity, PT, and Heptest with all formulations. The results indicate that PD 0313052 can be effectively administered subcutaneously to rabbits with these formulations and that alternative formulations of PD 0313052 can result in desirable changes in PK/PD profiles.