|
(a) |
(b) |
|
|
|
|
|
|


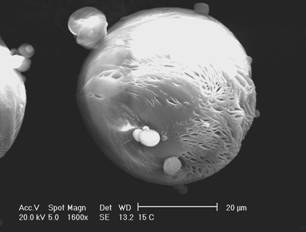
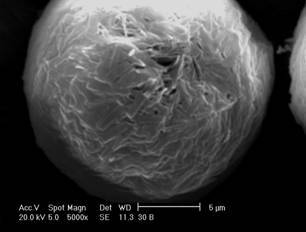
Figure 1:
Scanning
electron micrographs of CPT-loaded microspheres: (a) MC15, (b) MC30.
J Pharm Pharmaceut Sci (www.cspscanada.org) 9(1):22-31, 2006
Evaluation of antimetastatic activity and systemic toxicity of camptothecin-loaded microspheres in mice injected with B16-F10 melanoma cells
Cristiana Lima Dora1, Marcio Alvarez-Silva2, Andréa Gonçalves Trentin2, Tatiany Jovita de Faria1, Daniel Fernandes1, Robson da Costa1, Marco Stimamiglio2, Elenara Lemos-Senna1
1Laboratório de Farmacotécnica, Departamento de Ciências Farmacêuticas, Centro de Ciências da Saúde
2Laboratório de Neurobiologia e Hematologia Celular e Molecular, Centro de Ciências Biológicas; Universidade Federal de Santa Catarina, Campus Trindade, Florianópolis, Brazil
Received August 8, 2005; Revised December 5, 2005, Accepted December 7, 2005, Published December 22, 2005
Corresponding author: Elenara
Lemos-Senna,
Departamento de Ciências Farmacêuticas, Centro de Ciências
da Saúde,
Universidade Federal de Santa Catarina, Campus Trindade,
Florianópolis, 88040-970,
ABSTRACT Purpose: The aim of this work was to evaluate the
pulmonary antimetastatic activity and the systemic toxicity of
camptothecin-loaded microspheres. Methods: PCL microspheres containing camptothecin (CPT)
were prepared by the emulsion solvent/evaporation method and characterized
according to their encapsulation efficiency, particle size, morphology, and
drug release. The ability of CPT to inhibit the lung metastasis was verified
using an experimental mouse model intravenously injected with metastatic
B16-F10 melanoma cells. The microspheres and the free drug were given
intraperitoneally at a dose of 7 mg/kg at intervals of three or five days for
24 days. The systemic toxicity of CPT was evaluated by weight measurements,
survival and hemograms of the animals. Results: The encapsulation efficiency was nearly 80%. The
drug release was complete after 72 hours, but the burst effect increased from
7% to 35% with the increase in CPT content in the particles. It was observed
during the in vivo essays that all groups treated with CPT had a
decrease of nearly 70% in the number of lung metastases. However, systemic
toxicity was verified in animals that received the free drug. Conclusion: Camptothecin-loaded microspheres demonstrated
similar therapeutic efficacy when compared to those of the free drug, but the
toxicity was significantly reduced.
Camptothecin is the
alkaloid obtained from Camptotheca acuminata presenting a
considerable anticancer activity, in which the mechanism involves the
inhibition of topoisomerase I, an enzyme which is highly expressed in tumors.
This protein reduces the torsion stress of supercoiled DNA to facilitate the
replication, recombination and transcription processes. Camptothecin stabilizes
the normally transient DNA topoisomerase complex, leading to the cleavage of
doubled-strand DNA, and consequently, to cellular killing (1, 2). This drug is
widely distributed in the body, including the central nervous system, lungs,
liver and bowels (3). However, the use of camptothecin has shown some
drawbacks, which, in turn, have limited its application in therapeutics. This
drug encloses in its structure a highly conjugated pentacyclic ring with an a-hydroxylactone
portion at carbon 12 which is essential for its in vitro and in vivo antitumor activity
(4). At physiological pH, the lactone ring undergoes a rapid pH-dependent
non-enzymatic hydrolysis to form a less active and more toxic carboxylate form
(5). Stability studies in phosphate buffer pH 7.4 have demonstrated that
camptothecin’s half-life is about 10 minutes, and that only 13% of the drug is
found in lactone form at equilibrium. Therefore, the inactivation of this drug
occurs quickly a few minutes after intravenous administration. The inactivation
in plasma is further increased by preferential binding of the carboxylated form
to albumin that is about 200-fold over the lactone form (6). These drawbacks,
together with the poor water solubility conferred by the unusually weak basic
feature of its quinolone nitrogen atom, prevent the use of camptothecin by the
intravenous route (7). Several
hydrophilic derivatives have been developed in order to bypass the low aqueous
solubility of camptothecin. In spite of the water solubility improvements
provided by chemical modification, camptothecin derivatives approved for human
use, i.e. topotecan and irinotecan, are also susceptible to inactivation in a
physiological medium (7, 8, 9). Nevertheless, clinical trials have been carried
out with these camptothecin derivatives against a wide variety of tumors to
optimize administration schedules. So far, these studies have demonstrated that
large doses of camptothecins given intermittently are not effective. Camptothecins
require prolonged administration given continuously at low doses, or frequently
fractionated to produce a more effective antitumor activity (10). This can be
explained by the fact that topoisomerase I inhibitors exert their activity in
the S phase of the cell cycle. Hence, once the cytotoxic threshold is achieved,
the exposure time rather than the dose becomes the parameter which determines
antitumor activity. In addition, diarrhea and myelosuppression have been
reported as the most important dose-limiting toxicities of camptothecins (7, 11).
The severity of the undesirable effects of camptothecin is also dependent on
the administration schedule.
In view of the issues concerning low water solubility, poor stability and the need to maintain the CPT concentration in therapeutic levels at the tumor for prolonged time, there has been a considerable interest in the development of formulations that allow continuous delivery and the protection of camptothecin from inactivation in a physiological environment. Microspheres exhibiting prolonged release of a CPT derivative, the irinotecan (CPT-11), were firstly prepared by Machida et al. (1998) using poly(D,L-lactide) or poly(D,L-lactide-co-glycolide) (PLGA) as matrix. The CPT-11-loaded microspheres displayed marked antitumor activity against P388 ascitic tumor via intraperitonial administration; however, they were not significantly effective against Sarcoma 180 solid tumor implanted subcutaneously. Thus, the prolonged release of CPT-11 has shown to be effective in the i.p.-i.p. system but not in the i.p.-s.c. system (12). Furthermore, the in vivo studies have shown that therapeutic efficacy was better when the in vitro release rate from microspheres was higher (12). The PLGA microspheres were also proposed as a vehicle to stabilize the camptothecins inside the particles (13, 14). In fact, studies have shown that the acidic microclimate created from the hydrolysis of the PLGA into acidic oligomers and monomers favors the stabilization of CPT within the delivery device. The potential of local delivery of 10-hydroxycamptothecin-loaded PLGA micropheres in providing effective inductive chemotherapy was evaluated using a murine human oral squamous cell carcinoma regression model. In this study, PLGA microspheres showed significantly higher intratumor-drug concentrations relative to local bolus and i.p. administration (approximately 10 and 100 folder higher, respectively) leading to significant reduction of the tumor weight (15). Finally, in vitro cytotoxicity studies have demonstrated that CPT-loaded microspheres are more effective than free camptothecin against human derived RPMI-8402 lymphoid and THP-1 myeloid leukemia cell lines (16). More recently, in vitro cytotoxicity studies revealed that camptothecin encapsulated in PLGA microspheres retains its antitumor potency against B16 cells, being quickly uptaken by these cells (17). Even though many studies have been carried out involving the antitumor activity of microspheres incorporated with camptothecin, the improvement of the drug efficacy after systemic administration of the microspheres remains to be verified. In this study, then, we are interested in evaluating whether or not camptothecin microspheres are able to inhibit the growth and the lung metastatic spread in the mice intravenously injected with B16-F10 melanoma cells. These cells were obtained by the Fidler method which results in a cell line with increased pulmonary metastatic capacity (18). In our case, CPT microspheres were prepared and characterized using poly-e-caprolactone (PCL) as the polymer matrix former. Toxicity studies on microspheres and on the free drug were carried out after hemogram analysis and weight measurements of the mice. Since antimetastatic activity of camptothecin-loaded microspheres has not yet been described in detail in the literature, the present study attempts to shed light on this matter.
Camptothecin
and poly-e-caprolactone (MW 65000 Da) were obtained from
Sigma-Aldrich (USA). Hydroxypropylmethylcellulose (Methocel E4M Premium CR) was
purchased from Colorcon (USA). Except for methanol and acetonitrile used in
HPLC analysis (
A highly metastatic B16-F10 mouse
epithelial-like melanoma cell line was donated from Bio-Rio (
Camptothecin-loaded poly-e-caprolactone microspheres were prepared by an oil-in-water solvent emulsion/extraction technique. PCL (1g) was dissolved in 10 mL of methylene chloride solution containing 15 or 30 mg of camptothecin. This solution was gradually poured in 100 mL of aqueous phase containing hydroxypropylmethylcellulose 0.25% (w/v), which was previously saturated with the organic solvent. After the emulsion had been formed, 5 mL of ethanol were added and the mixture was kept under stirring at 500 rpm for five hours at room temperature. After the evaporation of the organic solvent, the hardened microspheres were centrifuged at 4000 rpm, washed three times with water, and freeze-dried. The microspheres prepared with initial amounts of 15 and 30 mg of camptothecin were denominated MC15 and MC30, respectively.
The camptothecin
content in the microspheres was determined by a reversed-phase HPLC method. The
analysis was carried out using a Supelcosil LC-18 column (15 cm x 4.6 mm ID, 5 mm; Supelco). The
mobile phase was composed of methanol: KH2PO4 10mM (50:50, v/v) adjusted at pH
2.8 with phosphoric acid, and it was delivered at flow rate of 1.0 mL/min. The
CPT was detected by UV absorption at 254 nm. Exactly weighed microspheres were
dissolved in a 1:1 methylene chloride:dimethyl
sulfoxide mixture and the resulting solutions were properly diluted with the
mobile phase prior to HPLC analysis. The samples were injected in triplicate
and the camptothecin concentration was determined by comparing the peak area
corresponding to the drug with that obtained with a standard camptothecin
solution. The encapsulation efficiency (%) was estimated as being the
percentage of camptothecin incorporated into the microspheres in relation to
the amount of drug initially added to the internal phase of the formulations.
The drug content was expressed as milligrams of camptothecin per 100 mg of
microspheres.
The morphological examination of the microspheres was carried out using a Philips XL30 scanning electron microscope (SEM) after coating the samples with gold under vacuum. After their dispersion in water, camptothecin-loaded microspheres were analyzed for their average size and size distribution using a laser diffraction analyzer (CILAS 1064, France) and were plotted for size distribution using the software supplied by the manufacturer.
In the release
studies, an amount of microspheres corresponding to 250
mg of camptothecin was exactly weighed and placed in 50
mL of a PBS pH 7.4 containing 2% (w/v) of Tween 80 in order to obtain sink
conditions. Samples were maintained
under stirring at 37ºC and at time intervals of 0, 1, 2, 4, 6, 10, 24, 48, 72
hours the microspheres were centrifuged and the supernatant was withdrawn and
frozen for further analysis. Camptothecin concentration in the release media
was determined by spectrofluorimetry. The solutions were excited at 374 nm and
the sample spectra were recorded in the wavelength region of 390 and 550 nm.
The samples’ emission spectrum areas were compared with those obtained with a
standard solution of camptothecin analyzed under the same conditions. The
analyses were carried out in triplicate and the camptothecin release (%) versus
time (hours) profiles were then plotted.
In the assays,
60-day-old Swiss male mice were used. The animals were maintained in a room (23
± 2oC and 60 ± 10% humidity) under a 12-hour light/dark cycle. Food and water
were given ad libitum. The in
vivo assays were previously approved by
our University’s Ethics Committee for Animal Use based on the Principles of
Animal Care.
Eight mice groups, each containing eight animals were employed in the evaluation of the antimetastatic activity of the microspheres. The mice were injected with 5 x 104 B16-F10 cells in 100 mL of PBS pH 7.4 via the intraorbital vein. Camptothecin-loaded microspheres, unloaded microspheres or the free drug were suspended in PBS pH 7.4 containing 0.3% (w/v) sodium carboxymethylcellulose and 0.2% (w/v) Tween 80, in order to improve the dispersion of the particles in the vehicle. On the second day after B16-F10 cells had been injected, the resulting suspensions were administrated intraperitoneally to the mice at a concentration of 7 mg/kg at intervals of three or five days, according to the experimental schedule tested.
Negative control only received the vehicle,
while the positive control received both the cells and the vehicle. The animals
were sacrificed in a CO2 chamber after 24 days. The lungs were
excised and fixed with a 10% formaldehyde solution and the metastatic colonies
were counted using a dissection microscope. The number of pulmonary metastasis
observed in the mice treated with the free drug, vehicle, unloaded microspheres
and camptothecin-loaded microspheres were compared. The statistical analysis
was performed using analysis of variance followed by the Bonferoni’s post-hoc,
using the Graph-Pad Prism (
The hematological toxicity of the free drug and of the camptothecin microspheres was evaluated using blood collected by cardiac puncturing in heparinized propylene tubes. Hemograms were obtained by the flow cytometry technique using a hematological Serono Baker System 9000 counter coupled with a Hematology Analyzer. The animals were weighted throughout the experiment and the dead ones were recorded; these data were also used as indicators of systemic toxicity.
In order to obtain microspheres with high
drug loading, formulations containing two initial amounts of camptothecin were
prepared. The results displayed in
Table 1 indicated that the encapsulation
efficiency of camptothecin was around 81% for both formulations.
Table 1: Camptothecin encapsulation values obtained
for MC15 and MC30 (n = 3).
|
|
Initial amount of CPT (mg) |
Encapsulation Efficiency (%)a
|
Drug content (%, w/w)b |
|
MC15 |
15 |
81.49 ± 8.95 |
1.17 ± 0.15 |
|
MC30 |
30 |
81.66 ± 3.94 |
2.37 ± 0.08 |
a weight of the encapsulated drug/ weight of total drug
used in preparation.
b
weight of encapsulated drug/ weight of microspheres.
However, the drug content increased from
1.17% to 2.37% (w/w) with the increase in the amount of
camptothecin initially added to the formulations. Several factors may affect
the encapsulation efficiency of the drug in the microparticles. In general,
high encapsulation values are observed when the
partitioning of the drug tends toward the internal phase of the emulsion, while
the fraction not encapsulated is eliminated in the filtration and washing
procedures (19). In view of this, the high values of encapsulation efficiency
obtained for camptothecin could be related to a higher
affinity of this hydrophobic drug for the internal phase of the emulsion. Similar
results were obtained when camptothecin was encapsulated in poly(D,L-lactic-co-glycolic
acid) microspheres, using the same drug to polymer ratio (14).
The morphology of microspheres was investigated by SEM. As
can be observed in the micrographs (Figure 1), the microencapsulation method
employed led to the formation of spherical particles with a rough surface.
This
surface characteristic may be related to rapid
methylene chloride removal resulting from the addition of ethanol to the
external phase during the evaporation process (20). The average diameter of
camptothecin-loaded microspheres was 32.64 and 40.49 mm for MC15 and MC30, respectively, and the size
distribution ranged from 0.4 and 120 mm
for both formulations.
The release profile of camptothecin from
both MC15 and MC30 was evaluated after suspension of the microspheres in PBS pH
7.4, containing 2% (w/v) Tween 80. The solubility of the drug in the release
medium was previously evaluated and it was found to be
79 mg/mL. Since the total amount of
camptothecin in the microspheres corresponded to 6.33% of its saturated
concentration, the perfect sink
conditions were reached. Spectrofluorimetry was used to determine the drug concentration in the release
medium because it is a very simple and rapid method. As can be observed in
Figure 2, nearly 100% of the drug was released after 72
hours for both MC15 and MC30.
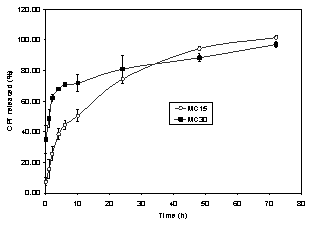
Figure 2: In vitro CPT release from PCL
microspheres in phosphate buffer solution pH 7.4
containing 2% (w/v) Tween 80 at 37° C.
However, the release profile was affected by the microsphere drug content. The initial
drug burst increased from 7 to 35% with the increment
in drug loading from 1.17 to 2.37 % for MC15 and MC30, respectively. Since all
formulation parameters remained constant, the higher burst effect obtained for
MC30 could be attributed to the presence of the fraction
of the associated drug at the particle surface (21). The burst effect
concomitantly increasing with camptothecin loading has been
reported when PLGA was used to prepare the microspheres, which is in
agreement with the release of lipophilic drugs from matrix systems (14).
The ability of
malignant neoplasms to produce secondary growths (metastases) in organs distant
from the primary tumors is the lethal event in the clinical course of most
neoplasic diseases. While primary cancers can be surgically
resected or locally irradiated, it is usually difficult to use these therapeutic
modalities against disseminated disease (18). The lungs and liver are the
earliest sites colonized by most metastatic tumors. Delivery of drugs is considered valuable not only for lung treatment, but also
for metastasis prevention even though the lung is not the metastatic target
organ (22). An important result of experimental metastasis models employing
direct injection of cells into the circulation has been the development of clonally
related variants that differ in metastatic potential.
Table 2: Effect of free drug and CPT-loaded
microspheres on pulmonary spontaneous metastasis in mice injected with 5x104
B16-F10 melanoma cells with different administration schedules (n = 8).
|
|
Dose and schedule |
Total dose
(mg/Kg) |
Number of pulmonary metastasis (M ±
s) |
Number of
deaths(a) |
|
Control (-) |
- |
- |
0 |
0 |
|
Control (+) |
- |
- |
22.38 ± 4.34 |
0 |
|
Unloaded microspheres |
With intervals of three days |
- |
20.35 ± 4.30 |
0 |
|
Unloaded microspheres |
With intervals of five days |
- |
18.63 ± 3.50 |
0 |
|
Free drug |
7 mg/kg with intervals of
three days |
42 |
Nd(b) |
8 |
|
Free drug |
7 mg/kg with intervals of
five days |
28 |
7.50 ± 1.69*** |
0 |
|
MC30 |
7 mg/kg with intervals of
three days |
42 |
6.37 ± 1.77*** |
0 |
|
MC30 |
7 mg/kg with intervals of
five days |
28 |
6.50 ± 3.12*** |
0 |
Statistical
significance was evaluated by analysis of variance
followed the Bonferoni’s post-hoc: *** p< 0.001 vs. positive and negative
control
(a) Total number of deaths after 24 days
(b) Nd – Not determined
A B16 melanoma cell line that is characterized by
progressively higher metastatic potential was developed by Fidler et al (23). While B16F1 parental cells are capable of forming experimental metastases
at a rate of ~1,3
´ 10-5 per cell per generation, the B16F10 cells generated by
successive tail vein metastasis had an effective metastasis rate of 5 x 10-5
per cells per generation. This experimental metastasis model provides several
advantages including the rapid time course for model maturity, the reproduction
and consistence of the biology of metastasis, and the control of the number of
cells that are introduced in the circulation. Some of
the disadvantages have been attributed to most of the experimental
metastasis models are chiefly related to the fact that the early steps in the
metastatic cascade are eliminated and the compressed time course of metastasis can
preclude their use in defining active agents against established metastatic
cancers (24). In spite of these disadvantages, several studies have emphasized
the interest in spontaneous lung metastasis model to evaluate the antitumor and
antimetastatic activity of drugs and drug delivery systems (22,25,26). Shao et al. (22), for instance, demonstrated that a cell-based drug delivery
system containing doxorubicin was more effective than the drug solution for both
the early treatment of metastasis and the eradication of established metastasis
in the lungs.
In this study, the effect of camptothecin-loaded
microspheres on spontaneous lung metastasis was verified
in mice inoculated via the retrorbital vein with B16-F10 cells. In this case,
the therapeutic efficacy of the microspheres was evaluated
for their ability to inhibit the number of lung colonies produced by the
B16-F10 cells injected by the intravenous route. MC30 was
selected for the in vivo
studies due to its higher drug content. Since the antitumor effectiveness of
camptothecin is highly dependent on the administration schedule (10), the
microspheres and the free drug were given intraperitoneally in the dose of 7
mg/kg, at intervals of either three or five days for 24 days. The number of
pulmonary metastasis and the number of deaths for each treated group are demonstrated in
Table 2.
The lungs with metastatic melanoma nodules produced after B16-F10 cell inoculation can be seen in Figure 3. We observed an increase in the melanoma nodules in positive controls as well as in the unloaded microparticles, whereas for all camptothecin schedules the amount of lesions was greatly diminished. As can be seen in Table 2, the number of lung metastases was significantly reduced after administration of the free drug at intervals of five days (± 64.5%), and after administration of MC30 at intervals of three (± 71.5%) or five days (± 71.0%), when compared to the number of metastases produced in animals used as positive controls (p < 0.001). All animals of the groups treated with camptothecin at intervals of 3 days died after 18 days, indicating the high drug toxicity with this administration schedule. In addition, the antimetastatic activity was similar in the groups treated with free camptothecin and microspheres.
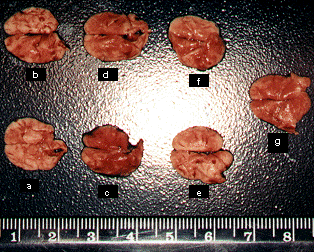
Figure 3: Appearance of the
lungs from mice injected intravenously with highly metastatic B16-F10 melanoma
cells (5x104) after treatment with (a) negative control; (b)
positive control; (c) unloaded microspheres given at intervals of three days;
(d) unloaded microspheres given at intervals of five days; (e) free camptotecin
given at intervals of five days; (f) MC30 given at intervals of three days and
(g) MC30 given at intervals of five days.
The administration
of camptothecin in prolonged and continuous schedules has shown that the
hematopoietic and mucosal progenitor cells with low topoisomerase I levels are
somewhat spared, while, more importantly, antitumor effects are maintained. In
these studies, myelosuppression caused mainly by severe neutropenia was found to be the dose-limiting toxic effect for most of
the tested protocols, but many other disturbances, such as hemorrhagic
cystitis, thrombocytopenia and diarrhea were also observed (27).
The weight measurements of the mice were performed during the experiments to evaluate the
systemic toxicity of camptothecin. At the end of the experiments, significant
weight loss of the animals was observed neither in the
negative and positive control groups, nor in the group that received the
unloaded and CPT-loaded microspheres at intervals of five days. However, when
the free drug was administered at intervals of 5 days,
and the MC30 was administered at intervals of three days, the weight of the
animals was significantly reduced as observed in
Figure 4 (p < 0.001).
Furthermore, the group that received free
camptothecin at intervals of three days developed severe diarrhea and did not
survive until the end of the experiments. Severe diarrhea have been
demonstrated to occur after consecutive daily injections of free drug and it
was considered to be an enterocolitis caused by the high level of CPTs retained
for a long period in the intestine (28).
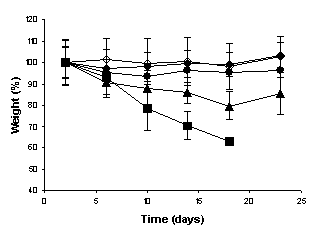
(a)
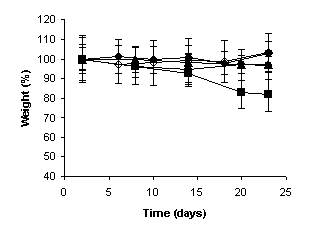
(b)
Figure
4: Percentage of weight loss of
animals injected intravenously with highly metastatic 5x104 B16-F10
melanoma cells and treated in administration schedules with intervals of (a)
three days and (b) five days over a 24-day period. (¨) negative control, (à) positive control, (l) Unloaded microspheres, (n) Free camptotecin, (p) MC30.
In order to compare
the toxicity produced by the administration of camptothecin in the free and
encapsulated forms, the hematological parameters of the animals were determined
on the twenty-fourth day, and were then compared with
the respective values obtained for normal control mice. The results of the
hemograms of the animals are shown in
Table 3.
Table 3: Effect of MC30 and free camptothecin (CPT) on
hematological parameters for each animal group after 24 days of treatment (n =
5).
|
|
Reference values |
Control |
Control
(+) |
MB(a) |
MB(b) |
CPT(b) |
MC30(a) |
MC30(b) |
|
|
|
|
|
|
|
|
|
|
|
Total RBC Count (x 106/mm3) |
7.0 -12.5 |
8.12 ± |
8.21 ± |
8.74 ± |
8.51 ±
|
7.15 ±
|
7.25 ± |
6.96 ± |
|
Hemoglobin (g/dL) |
10.2 -16.6 |
13.73 ± |
14.07 ±
0.79 |
14.46 ±
0.73 |
14.50 ±
0.66 |
11.13 ±
|
12.87±
|
10.45 ± |
|
Hematocrit (%) |
39 - 49 |
40.96 ± |
41.22 ±
3.80 |
42.73 ±
1.55 |
42.80 ±
2.80 |
34.73 ±
|
36.45 ±
1.80 |
31.82 ± |
|
White Blood cells |
|
|
|
|
|
|
|
|
|
Total WBC Count (mm3) |
6000 - |
10133.33 ± 1556.70 |
9100 ±
4573.11 |
9733.33 ± 2968.3 |
9675± 3525.50 |
8266.67± 3074.63 |
10825± 5483.54 |
7112.50± 2172.7 |
|
Neutrophil (%) |
10 -40 |
17.00± |
15.75±
6.30 |
24.66±
13.58 |
25.50±
15.02 |
2.33± |
24.33± 10.41 |
13.25 ± |
|
Eosinophil (%) |
0 - 4 |
0 |
1 |
0 |
0 |
0 |
0 |
0 |
|
Lymphocyte (%) |
55 - 95 |
85.66 ± |
82.25 ± 7.09 |
74.6 ±
14.15 |
73.75±
14.93 |
97.33± |
79.75± 12.58 |
86.50 ± |
|
Monocyte (%) |
0.1 - 3.5 |
1.5 ± 0.7 |
2.33 ±
1.15 |
2 |
1 |
1 |
1 |
1 |
|
Platelet
|
0.8 - 1.1 |
1.01 ± 0,07 |
1.11 ±
0.13 |
1.49 ±
0.20 |
1.35±
0.22 |
1.82± 0.23 |
1.7± 0.04 |
1.8 ± 0.16 |
(a) Given at intervals of three days
(total dose = 42 mg/Kg).
(b) Given at intervals of five days
(total dose = 28 mg/Kg).
The
count of red blood cells, hemoglobin content, and hematocrit values were
slightly lower in the groups that received the free drug and the MC30
microspheres. These results can be related to some
toxicity induced by the camptothecin and not by the tumor, since the positive
control group did not show a reduction of these elements. Surprisingly, after
the administration of camptothecin there was an increase in the count of
platelets (thrombocytosis), rather than thrombocytopenia. Thrombocytosis was,
nevertheless, also verified after the administration of the unloaded
microspheres, suggesting that this may be a consequence of an inflammatory
processes caused by the intraperitoneal administration of microparticles in
suspension.
On
the other hand, neutropenia was found in the group of
mice treated with free camptothecin at intervals of 5 days. In groups that
received the MC30 microspheres, at intervals of 3 or 5
days, this effect was not observed, which, in turn, indicates a decrease in
hematological toxicity when the encapsulated drug is administered.
The pharmacokinetics of camptothecin has been described elsewhere. Under physiological conditions, camptothecin exists in equilibrium between its lactone (CPT) and carboxylate forms (CPT-Na), this equilibrium favoring the carboxylate form. The activity of CPT was found to be approximately 10-fold greater than Na-CPT and this was attributed to a small amount of in vivo conversion from carboxylate to lactone (29). Furthermore, the distribution clearance for lactone was greater than the carboxylate form, indicating that the lactone is quickly distributed into the tissue compartment (30). In comparative pharmacokinetics studies performed in rats, the plasma concentration of irinotecan, a water soluble camptothecin derivative, was gradually increased when microspheres were administrated by i.p route, while a quick decreasing on the plasma concentration of the drug was observed after the administration of the drug solution (31). In our case, since the same dose was given when either microspheres or CPT dispersion were administered, the reduction in the systemic toxicity can be associated with both the protection of the drug inside of the particles, avoiding its conversion in the carboxylate form, and the slower delivery of lactone form towards the blood stream. Therefore, it is possible that the administration of microspheres provides lower CPT plasma concentration in relation to the drug dispersion, contributing to the toxicity reduction but maintaining the antimetastatic effect.
Camptothecin-loaded microspheres displayed similar antimetastatic activity as compared to the free drug, but their systemic toxicity was lower as evidenced by the evaluation of the weight loss and survival of the animals, and blood neutrophil count. The decrease of toxicity may be related to the maintenance of the camptothecin lactone ring in the microspheres, which leads the drug to achieve systemic circulation in lower concentrations than when it is administrated in its free form. The hematological toxicity reduction was observed for both administration schedules when the drug was encapsulates in PCL microspheres, indicating the microencapsulation of CPTs can be advantageous to minimize the inconvenient of the systemic administration of these drugs.
The authors wish to thank CNPq for financial support.
[1]
Hsiang, Y.H., Hertzberg, R., Hecht S. and
Liu, L., Camptothecin induces protein-linked DNA breaks via mammalian
[2] Iyer,
L. and Ratain, M.J., Clinical pharmacology of camptothecins. Cancer Chemother. Pharmacol. 42:31-43, 1998.
[3] Potmesil,
M. Camptothecins: from bench research to hodpital wards. Cancer Research. 54:
1431-1439, 1994.
[4]
[5] Fassberg,
J., Stella, V.J., A kinetic and mechanistic study of the hydrolysis of
camptothecin and some analogues. J.
Pharm. Sci. 81:676-684, 1992.
[6] Burke,
T.G. and Mi, Z., The structural basis of camptothecin interactions with human
serum albumin: impact on drug stability. J. Med. Chem. 37:40-46, 1994.
[7] Garcia-Carbonero,
R. and Supko, J., Current perspectives on the clinical experience,
pharmacology, and continued development of the camptothecins. Clin.
Cancer Res. 8:641-661, 2002.
[8] Herben,
U.M.M, Bockkel, W.W., Scheilens, J.H.M and Beijen, J.H., Clinical
pharmacokinetics of camptothecin topoisomerase I inhibitors. Pharm. World Sci. 20 161-171, 1988.
[9] Hatefi, A. and Amsden, B., Camptothecin
delivery methods. Pharm.
Res. 19: 1389-1399, 2002.
[10] Thompson, J., Stewart, C. and Houghton, P.,
Animal models for studying the action of topoisomerase I targeted drugs. Biochim. Biophys.
Acta, 1400: 301-319, 1998.
[11] O’Leary,
J. and Muggia, F.M., Camptothecins: a review of their development and schedules
of administration. Eur. J. Cancer. 34: 1500-1508, 1998.
[12]
[13] Shenderova, A., Burke, T.G. and Schwendeman, S.P., Stabilization of
10-hydroxycampthotecin in poly(lactide-co-glycolide) microspheres delivery
vehicles. Pharm. Res., 14(10): 1406-1410, 1997.
[14] Ertl, B., Platzer, P., Wirth, M. and Gabor,
F., Poly (DL-lactide-co-glycolide acid) microspheres for sustained delivery and
stabilization of camptothecin. J. Control. Rel., 61(3):305-317, 1999.
[15] Mallery, S.R., Shenderova, A., Pei, P., Begum, S., Chimineri,
J.R., Wilson, R.F., Castro, B.C., Schuller, D.E. and Morse, M.A., Effects of
10-hydroxycamptothecin delivery from locally injectable
poly(lactide-co-glycolide) microspheres in a murine human oral squamous cell
carcinoma regression model. Anticancer Res.,
21(3B):1713-1722, 2001.
[16] Kumar, V., Kang, J.and Hohl, R.J., Improved dissolution and
citotoxicity of camptothecin incorporated into oxidized-cellulose microspheres
prepared by spray-drying. Pharm. Dev. Technol.,
6(3): 459-467, 2001.
[17] Tong, W., Wang, L., D’Souza, M.J.,
Evaluation of PLGA microspheres as delivery for antitumor agent-camptothecin. Drug.
[18] Fidler,
I.J. Origin and biology of cancer metastasis. Cytometry. 10:673-680, 1990.
[19]
[20] Bodmeier,
R. and McGinty, J.W., The preparation and evaluation of drug-containing poly
(dl-lactide) microspheres formed by the solvent evaporation method. Pharm. Res., 4:465-471, 1987.
[21] Huang, X. and Brazel, C., On the importance and mechanisms of burst release in
matrix-controlled drug delivery systems. J. Control. Rel. 73:121-136,
2001.
[22] Shao, J., DeHaven, J.,Lamm,
D., Weissman, D.N., Malanga, C.J., Rojanasakul, Y., Ma, J.K.H. A Cell-based drug delivery System for lung targeting: II
Therapeutic Activities on B16-F10 Melanoma in Mouse Lungs. Drug Delivery. 8:71-76, 2001.
[23] Poste, G., Doll, J., Hart,
I.R., Fidler, I.J. In vitro selection of murine B16 melanoma
variants with enhanced tissue-invasive properties. Cancer.
Res. 40:1636-1644, 1980.
[24] Khanna, C., Hunter, K.
Modeling metastasis in vitro. Carcinogenesis.
26(3):513-523, 2005.
[25] Chirivi, R., Garofalo, A., Crimmin, M.J.,
Bawden, L. A. Stoppacciaro, P.D. Brown, R. Giavazzi, Inhibition of the
metastatic spread and growth of B16-BL6 murine melanoma by synthetic matrix metalloproteinase
inhibitor. Int. J. Cancer. 58: 460-464, 1994.
[26] Yoshikawa, N., Nakamura, K., Yamaguchi, Y.,
Kagota, S., Sinozuka, K., and Kunitomo, M., Effect of PKC412, a selective
inhibitor of protein kinase C, on lung metastasis in mice injected with B16
melanoma cells. Life Sciences. 72:1377-1387, 2003.
[27] Slichenmyer,
W. and von Hoff, V., New natural products in cancer chemotherapy. J. Clin. Pharmacol., 30: 770-788, 1990.
[28] Araki, E., Ishikawa, M., Iigo, M., Koide, T., Itabashi, M. and
Hoshi, A. Relationship between development of diarrhea and the concentration of
SN-38, an active metabolite of CPT-11, un the
intestine and the blood plasma of athymic mice following intraperitonial
administration of CPT-11. Jap. J. Cancer.
Res., 84(6): 607-702, 1993.
[29] Hertzberg, R. P., Caranfa, M.J., Holden, K.G.., Jakas, D.R.,
Gallagher, G., Mattern, M.R., Mong, S.M., Bartus, J.O., Johnson, R.K.,
Kingsbury, W.D. Modification of hydroxyl lactone ring of camptothecin;
Inhibition of mammalian Topoisomerase I and biological activity. J. Med.
Chem. 32:715-720, 1989.
[30] Scott, D.O., Bindra, D.S., Stella, V.J.
Plasma Pharmacokinetics of Lactone and Carboxylate Forms of 20(S)-Camptothecin
in Anestherized Rats. Phar.
Res., 10(10):1451-1457, 1993.
[31]
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.