J Pharm Pharmaceut Sci
(www.cspscanada.org) 9(1):10-21, 2006
Effects of water deprivation on the pharmacokinetics
of DA-8159, a new erectogenic, in rats
Ji Young Kim1, Yu Chul Kim1,
Myung Gull Lee1,
Jong Won Kwon2, Moohi Yoo2
1College of Pharmacy and Research
Institute of Pharmaceutical Sciences,
Seoul
National University,
Seoul, South Korea
2Research Laboratory,
Dong-A
Pharmaceutical Company, Ltd., Yongin,
South Korea
Received July 26, 2005; Revised September 19,
2005; Accepted October 26, 2005; Published December 21, 2005
PDF Version
ABSTRACT.
Purpose: To test the
effect of 72 h water deprivation on the non-renal clearance (CL) of DA-8159 in a
rat model of dehydration. DA-8159 is mainly metabolized via CYP3A1/2 and the
expression and mRNA level of CYP3A1/2 are not affected by dehydration. Methods:
DA-8159 (30 mg/kg) was administered intravenously or orally to male control
Sprague–Dawley rats and rat model of dehydration. Results: As expected, after intravenous administration, the CLNR
values of DA-8159 were comparable between two groups of rats. This could be
supported by comparable intrinsic CL of DA-8159 using hepatic microsomes
for both groups of rats. However, the
CL was significantly slower in rat model of dehydration due, at least in part, to
significantly slower renal CL in rat model of dehydration. The slower CLR
in rat model of dehydration could be due to urine flow rate-dependent renal CL
of DA-8159; the less urine output, the less the urinary excretion of unchanged
DA-8159. After oral administration, the AUC values of DA-8159 were not significantly different between two groups
of rats, although the AUC of DA-8159 in rat model of dehydration was
significantly greater than controls after intravenous administration. This
could be possibly due to changes in the intestinal first-pass effects in rat
model of dehydration. Conclusions:
After intravenous administration of DA-8159, the non-renal CL values were
comparable between two groups of rats due to the lack of effect of dehydration on
CYP3A1/2.
INTRODUCTION
DA-8159 (Udenafil), 5-[2-propyloxy-5-(1-methyl-2-pyrollidinylethylamidosulfonyl)phenyl]-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo(4,3-d)pyrimidine-7-one,
a new inhibitor of cyclic guanosine monophosphate (cGMP)-specific
phophodiesterase type V (PDE V), has been synthesized for the treatment of male
erectile dysfunction (Research Laboratory of Dong-A Pharmaceutical Company,
Yongin, South Korea). DA-8159 is metabolized to DA-8164 (N-dealkylated DA-8159:
5-[2-propyloxy-5-(aminosulfonyl) phenyl]-1-methyl-3-propyl-1,
6-dihydro-7H-pyrazolo (4, 3-d) pyrimidine-7-one) in mice, rats, rabbits, dogs,
and humans (1). Mechanism (2) and erectogenic effects (3, 4) of DA-8159 have
been reported. DA-8159 is a potent, selective, and competitive inhibitor of
human PDE V. In vitro experiments
using a series of PDE isozymes (PDE I, II, III, V, and VI) indicated that
DA-8159 is highly selective and potent antagonist of PDE V from human and
rabbit platelets producing IC50 values of 8.25 and 5.84 nM,
respectively (2). Oral DA-8159 is now being evaluated in phase III clinical
trial for the treatment of male erectile dysfunction in Korea (Dong-A Pharmaceutical Company, Seoul, South
Korea).
Recently, Kim et al. (5) have reported that
metabolism of DA-8159 and formation of DA-8164 is mainly catalyzed via the hepatic
microsomal cytochrome P450 (CYP) 3A1/2 in male Sprague Dawley rats. For
example, pretreated with dexamethasone (a main inducer of CYP3A1/2 in rats),
reduced the area under the plasma concentration–time curve (AUC) of DA-8159 after
intravenous administration to male Sprague–Dawley rats while increased that of
DA-8164 is increased. On the other hand, pretreated with troleandomycin (a main
inhibitor of CYP3A1/2 in rats) has opposite effects on the AUC of the drug and
its metabolite. However, 3-methylcholanthrene, phenobarbital, and isoniazid (main
inducers of CYP1A1/2, 2B1/2, and 2E1, respectively, in rats), and quinine (a
main inhibitor of CYP2D1 in rats) appears to have no effect of the AUC of
DA-8159.
Dehydration occurs by
excessive sweating, polyuria, severe diarrhea, and hyperthermia (6). Water
deprivation may cause significant hormonal, physiological, and biochemical
changes in the body (7–11 and references therein). For example, kidney and/or
liver functions seemed to be impaired in rat model of dehydration based on
blood and urine chemistry data and/or microscopic examinations of the kidney
and liver. Therefore, it could be expected that the pharmacokinetics and hence
pharmacodynamics of drugs could be altered in water deprivation. Since, the
first report on the effects of water deprivation on aspirin disposition
kinetics (6), water deprivation has been reported to alter the disposition
kinetics of various drugs (7–11 and references therein).
Kim et al. (12) reported that
in male Sprague–Dawley rats with 72-h water deprivation (rat model of
dehydration), the expressions of CYP1A2, 2B1/2, 2C11, and 3A1/2 were not
changed, however, that of CYP2E1 was three-fold induced based on Western blot
analysis. The mRNA level of CYP2E1 also increased in rat model of dehydration
based on Northern blot analysis (12). Hence, it would be expected that the
hepatic metabolism of DA-8159 would not changed in rat model of dehydration.
Although the pharmacokinetic changes of many drugs in rat model of dehydration were
reported (7–11 and references therein), the changes with respect to CYP isozymes
seemed not to be reported except chlorzoxazone (11) and oltipraz (13). In this
regard, DA-8159 was chosen in this study, since the drug is metabolized mainly
via CYP3A1/2 in rats (5) and CYP3A1/2 was not changed in rat model of
dehydration (12). Moreover, DA-8159 could be used in humans with dehydration state.
The purpose this study is to find whether the pharmacokinetic parameters of
DA-8159 are not changed after intravenous and oral administration of DA-8159 in
rat model of dehydration with respect to CYP3A1/2.
METHODS
AND
MATERIALS
Chemicals
DA-8159, DA-8164, and sildenafil [an internal
standard of high-performance liquid chromatographic (HPLC) analysis of DA-8159
and DA-8164] were supplied from Research Laboratory of Dong-A Pharmaceutical
Company. N,N-dimethylacetamide (DMA), b-nicotinamide
adenine dinucleotide phosphate (NADPH; as a tetrasodium salt), ethylenediamine tetraacetatic acid
(EDTA), and tri(hydroxymethyl)aminomethane
(Tris)-buffer were products from Sigma–Aldrich
Corporation (St. Louis, MO). Polyethylene glycol 400 (PEG 400) was purchased from Duksan Chemical
Company (Seoul, South Korea). Other chemicals were
of reagent grade or HPLC grade.
Animals
Male Sprague–Dawley
rats (weighting 270 to 315 g) were purchased from Charles River Company Korea (Orient, Seoul, South Korea).
All rats were maintained in a light-controlled room (light: 0700–1900, dark:
1900–0700) kept at a temperature of 22 ± 2 °C and a relative humidity of 55 ± 5% (Animal Center for Pharmaceutical
Research, College of Pharmacy, Seoul National University, Seoul, South Korea). The rats were randomly divided
into two groups, control rats and rat model of dehydration. For control rats,
water and food (Sam Yang Company, Seoul,
South Korea)
were supplied ad libitum for 72-h; for rat model of dehydration, water
was deprived for 72-h with free access to food. Food intakes and body weights
were recorded daily for four days (before water deprivation and the first,
second, and third days after water deprivation) for each group of rats. The study
protocol was approved by the Animal Care and Use Committee of the Collage of
Pharmacy, Seoul National University.
Measurement of Vmax, Km, and CLint
for the Disappearance of DA-8159 and for the Formation of DA-8164 in Hepatic Microsomal
Fractions
The procedures were similar to the reported methods (11). The livers of
control rats and rat model of dehydration (n
= 6; each) were homogenized (Ultra-Turrax T25; Janke and Kunkel,
IKA-Labortechnik, Staufeni,
Germany). The
protein level was measured by the reported method (14). The Vmax (the maximum velocity)
and Km (the
Michaelis–Menten constant; the concentration at which the rate is one-half of Vmax) for the disappearance
of DA-8159 and for the formation of DA-8164
were determined after incubating the above microsomal fraction
(equivalent to 1 mg of protein), a 10-mL aliquot
of DA-8159 (dissolved in 0.05 M citric acid to have substrate concentrations of
1, 2, 5, 10, 20, 50, and 200 mM), and a
50-mL (1.2 mM) aliquot of NADPH in a total
volume of 300 mL by 100 mM phosphate buffer (pH 7.0), in a
water-bath shaker kept at 37 oC and at a rate of 50 oscillations per
min (opm) for 10 min. All of the above microsomal incubation conditions were
linear. The reaction was terminated by adding a 1-mL aliquot of ethylether after
10-min incubation, and a 100-mL aliquot of 0.1 N Na2CO3 containing 3 mg/mL
of sildenafil (an internal standard) was added. DA-8159 and DA-8164 were
measured by the reported HPLC method (15). The kinetic constants (Km and Vmax) for the disappearance of
DA-8159 and for the formation of DA-8164 were calculated using the Lineweaver–Burk
plot (16) with the method of least squares. The intrinsic clearance (CLint) values for the
disappearance of DA-8159 and for the formation of DA-8164 were calculated by
dividing respective Vmax
by respective Km.
Intravenous Administration
of DA-8159 or DA-8164 to Rats
On the fourth day, the carotid artery (for
blood sampling) and the jugular vein (for drug administration) of each rat were
cannulated under the light ether anesthesia (17,18). And
then, each rat was housed
individually in a rat metabolic cage (Daejong Scientific Company, Seoul, South
Korea) and allowed for 4–5 h to recover from
the anesthesia before the study began. DA-8159 (dissolved in 0.05 M citric
acid) at a dose of 30 mg/kg was
infused (total infusion volume of approximately 0.6 mL) over 1-min via the
jugular vein of control rat (n = 9)
and rat model of dehydration (n = 10).
Approximately 0.22-mL aliquot of blood sample was collected via the carotid
artery at 0 (to serve as a control), 1 (at the end of the infusion), 5, 15, 30,
60, 120, 180, 240, 360, 480, 600, and 720 min after intravenous administration of DA-8159. Heparinized 0.9%
NaCl-injectable solution (20 units/mL), approximately
0.3 ml, was used to flush each cannula after each blood sampling to prevent
blood clotting. At the end of experiment (24 h), each metabolic cage was rinsed
with 15 mL of distilled water and the rinsings were combined with 24-h urine
sample. After measuring the exact volume of combined urine sample, a 0.1-mL
aliquot of the combined urine sample was stored in a −70°C freezer (Revco
ULT 1490 D-N-S; Western Mednics, Asheville,
NC) until HPLC analysis of DA-8159 (15). At the same time (24 h), as much blood
as possible was collected via the carotid artery and each rat was sacrificed by
cervical dislocation. And then, the entire gastrointestinal tract (including
its contents and feces) was removed, transferred into a beaker containing 100 mL
of methanol (to facilitate the extraction of DA-8159 and DA-8164) and cut into
small pieces using scissors. After stirring with a glass rod for 1 min, two 0.1-mL
aliquots of the supernatant were collected and stored in a –70°C freezer until HPLC analysis of DA-8159
and DA-8164 (15). Similar experiment was also performed with DA-8164. DA-8164
(dissolved in DMA : PEG 400 = 1 : 1; v/v, to produce a
concentration of 5 mg/mL) at a
dose of 10 mg/kg was infused to
control rats (n = 9) and rat model of
dehydration (n = 9). Approximately
0.22-mL aliquot of blood sample was collected via the carotid artery at 0, 1,
5, 15, 30, 60, 120, 180, 240, 360, 480, 600, and 720 min after intravenous
administration of DA-8164. Other procedures are similar to those of DA-8159
studies.
Oral Administration of DA-8159 in Rats
On the fourth day, the carotid artery of
each rat was cannulated under light ether anesthesia (17,18).
DA-8159 (the same solution as used in the intravenous study) at a dose of 30 mg/kg was administered
orally using a feeding tube in control rats (n = 11) and rat model of dehydration (n = 10). Blood samples were collected
at 0, 15, 30, 45, 60, 90, 120, 180, 240, 360, 480, 600, or 1440 min after oral
administration of DA-8159. Other procedures were similar to those in the intravenous
studies.
Protein Binding
of DA-8159 to Rat Plasma Using an Equilibrium Dialysis Technique
Plasma protein binding of DA-8159 to additional control rats and rat
model of dehydration (n = 5; each) was
determined at a DA-8159 plasma concentration of 5 mg/mL using an equilibrium dialysis technique (19). One
mL of fresh plasma was dialyzed against 1 mL of isotonic Sørensen phosphate
buffer (pH 7.4) containing 3% dextran, with 1 mL dialysis cell (Fisher
Scientific, Fair Lawn, NJ)
and Spectra/Por 4 membrane (mol. wt. cutoff of 12,000–14,000; Spectrum Medical
Industries, Los Angeles, CA). The spiked dialysis cell was incubated
for 24 h in a water-bath shaker kept at 37 oC and at a rate of 50
opm (19).
HPLC Analysis
of DA-8159 and DA-8164
The concentrations of DA-8159 and DA-8164 in the above biological
samples were analyzed by the slight modification of the reported HPLC method (15).
To a 0.1-mL aliquot of biological sample, a 0.1-mL aliquot of 0.1 N Na2CO3 containing 3 mg/mL of sildenafil (an internal standard)
and a 1-mL aliquot of ethylether were added. After vortex-centrifugation at 16,000
g for 2 min, the ether layer was
collected and dried under a gentle stream of nitrogen gas. A 0.1-mL aliquot of the mobile phase was added
to reconstitute the residue and a 0.05-mL aliquot was injected directly onto a
reversed-phase (C18) column. The mobile phase, 20 mM KH2PO4
(pH = 4.7) : acetonitrile (72 : 28; v/v), was run at a
flow-rate of 1.5 mL/min and the column effluent was monitored by an UV detector
set at 292 nm at room temperature. The retention times of DA-8159, DA-8164, and
sildenafil were approximately 9.7, 17.1, and 6.9 min, respectively. The
detection limits of DA-8159 and DA-8164 in plasma and urine were all 0.02 mg/mL. The coefficients of variation were below
9.4%.
DISCUSSION
Induction of dehydration was evident in rat model of dehydration; body
weight gain and 24-h urine output were significantly smaller and hematocrit was
significantly greater than controls (Tables 2–4). After intravenous
administration in control rats, body weight gain increased with days; the mean
body weights were 303 ± 28.9, 316 ± 18.9, 319 ± 17.0, and 320 ± 14.6 g for before and the first, second,
and third days, respectively. However, in rat model of dehydration, dehydration
caused significant decrease in body weight gain; the corresponding values were 310
± 28.7, 287 ± 22.6, 274 ± 23.1, and 253
± 24.2 g. In
control rats, daily food intakes were almost constant; the mean values were 22.1
± 4.40, 26.1 ± 4.79, and
25.1 ± 1.08 g for the first, second, and third days,
respectively. However, in rat model of dehydration, food intakes decreased with
days; the corresponding values were 15.7 ± 5.07, 8.21 ± 2.61, and 5.36 ± 2.01 g. Similar results were also obtained
after oral administration (data not shown). The above data indicated that
significant decrease in body weight gain in rat model of dehydration was due to
less food consumption in addition to water deprivation. Similar results were
also reported from other rat studies (11,12).
DA-8164 was a main
metabolite in humans and pharmacological effect of DA-8164 in terms of PDE V
inhibitory activity was half of that of DA-8159 (an internal report). Hence, the
pharmacokinetics of DA-8164 was evaluated in this study. Shim et al. (18) reported
that the AUC values of DA-8159 were dose-proportional after intravenous administration
at doses of 5–30 mg/kg and oral administration at doses of 20–30 mg/kg in rats.
Hence, the 30 mg/kg of DA-8159 was arbitrarily chosen in this study.
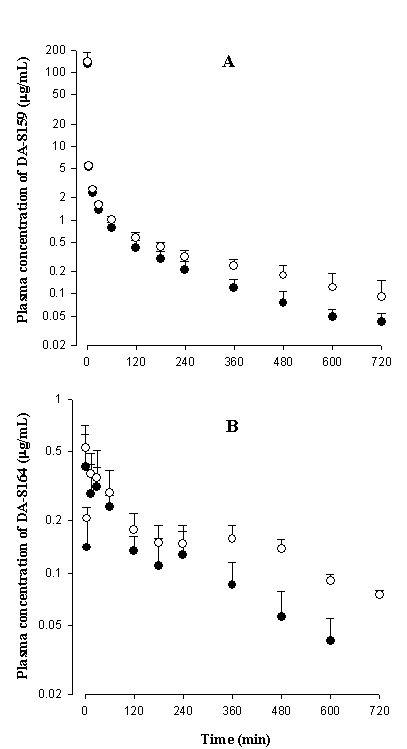
After intravenous
administration of DA-8159, contribution of CLR to CL of DA-8159 was
not considerable; the Ae0–24 h, DA-8159 values were less than 7.79% of
intravenous dose of DA-8159 for both groups of rats (Table 2), indicating that
most of the intravenously administered DA-8159 are eliminated via the nonrenal
route (CLNR). The contribution of gastrointestinal (including
biliary) excretion of unchanged DA-8159 to CLNR of DA-8159 seemed
also not to be considerable; the GI24 h, DA-8159 values were less
than 1.97% of intravenous dose of DA-8159 for both groups of rats (Table 2). The
small value in GI24 h, DA-8159, less than 1.97%, could not
be due to chemical degradation of DA-8159 in gastrointestinal tract; DA-8159
was stable in various pH solutions (25). Moreover, the percentages of oral dose
of DA-8159 (30 mg/kg) excreted in 24-h bile as unchanged drug were < 0.1% in
4 rats (18). The above data indicated that the CLNR values of
DA-8159 listed in Table 2 could represent metabolic clearances of DA-8159 in
rats.
After intravenous administration
of DA-8159, the CLNR values of DA-8159 were comparable between two
groups of rats (Table 2), and this could be expected because DA-8159 was mainly
metabolized via CYP3A1/2 in rats (5) and CYP3A1/2 was not changed in rat model
of dehydration (12). This could be supported by comparable in vitro CLint values for the disappearance of DA-8159
for both groups of rats (Table 1). After intravenous administration of DA-8159
in rat model of dehydration, the significantly slower CL of DA-8159 was at
least partly due to significantly slower CLR of DA-8159 in rat model
of dehydration, although the contribution of CLR of DA-8159 were not
considerable as mentioned earlier (Table 2). The significantly longer MRT of
DA-8159 in rat model of dehydration could support the slower CL of DA-8159 in rat
model of dehydration (Table 2). The slower CLR of DA-8159 in rat
model of dehydration could be mainly resulted from significantly smaller Ae0-24
h, DA-8159 and
significantly greater AUC of DA-8159 in rat model of dehydration (Table 2). The
smaller Ae0-24
h, DA-8159 in rat model
of dehydration could be due to urine flow rate-dependent CLR of
DA-8159 in rats. Recently, Kim et al. (26) reported that CLR of
DA-8159 was dependent on urine flow rate in rats; the less urine output, the
less Ae0-24
h, DA-8159. The 24-h
urine output was significantly smaller in rat model of dehydration than
controls (93.1, 89.1, and 70.3% decrease; Tables 2–4). The smaller Ae0-24
h of DA-8159 in rat
model of dehydration may also be due to impaired kidney function in rat model
of dehydration. Impaired kidney function in rat model of dehydration was also reported
from other rat studies (7–11 and references therein).
After intravenous
administration of DA-8159 in rat model of dehydration, the Vdss of DA-8159 was significantly
larger than controls (Table 2). However, this could not be due to increase in
free (unbound to plasma proteins) fractions of DA-8159 in plasma in rat
model of dehydration. The plasma protein
binding values of DA-8159 were 67.2 ± 1.57 and 70.2 ± 3.82% for
control rats and rat model of
dehydration, respectively; they were
not significantly different.
After intravenous administration of DA-8159 in rat model of dehydration,
the AUC of DA-8164 was significantly greater than controls (Table 2). However, this
was not due to increase in expression of CYP3A1/2 in rat model of dehydration since
CYP3A1/2 was not changed in rat model of dehydration (12). This could be
supported by comparable in vitro CLint
values for the formation of DA-8164 for both groups of rats (Table 1). The
greater AUC of DA-8164 could be due to greater exposure of the parent drug (the
significantly greater AUC of DA-8159) in rat model of dehydration (Table 2). In
order to explain the significantly greater AUC of DA-8164 after intravenous
administration of DA-8159 in rat model of dehydration, DA-8164 was administered
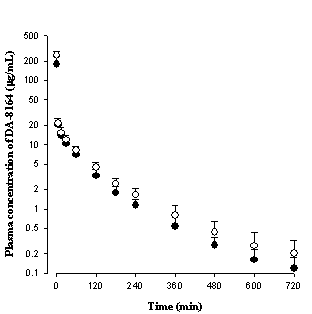
intravenously in both groups of rats. After intravenous administration of
DA-8164 in rat model of dehydration, the AUC of DA-8164 was significantly
greater than controls (Table 3). Moreover, the CL of DA-8164 (Table 3) was
significantly slower than that of DA-8159 (Table 2). This factor could also
contribute to the significantly greater AUC of DA-8164 after intravenous
administration of DA-8159 in rat model of dehydration (Table 3).
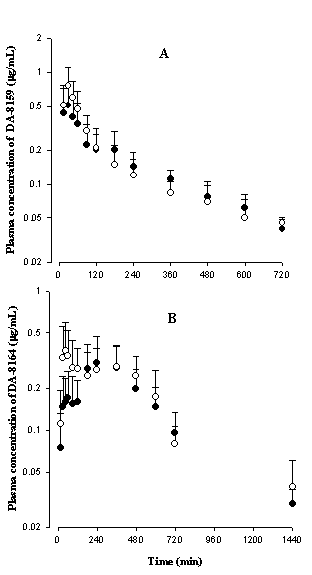
After intravenous administration
of DA-8159 in rat model of dehydration, the AUC of DA-8159 was significantly
greater than controls (Table 2). However, after oral administration of DA-8159,
the AUC of DA-8159 was comparable between two groups of rats (Table 4). However,
this was not due to decrease in gastrointestinal absorption of DA-8159 in rat
model of dehydration. Based on the linear pharmacokinetics (18), the mean “true”
fractions of oral dose unabsorbed (Funabs)
in this study were estimated based on the reported equation (27). The estimated
Funabs values
were 1.35 and 1.13% for control rats and rat model of dehydration,
respectively. Hence, more than 98% of oral dose of DA-8159 were absorbed from
the gastrointestinal tract for both groups of rats. The comparable AUC values
of DA-8159 after oral administration of DA-8159 could be due to changes in
first-pass effect in rat model of dehydration; the intestinal first-pass effect
of DA-8159 at a dose of 30 mg/kg was approximately 59% of oral dose in rats (18).
After oral administration of DA-8159, formation of DA-8164 increased compared
with that after intravenous administration; the AUCDA-8164/AUCDA-8159
ratios after intravenous administration of DA-8159 were 18.1 and 28.1% for
control rats and rat model of dehydration, respectively, however, the
corresponding values after oral administration of DA-8159 were 173 and 206%. This
could be due to considerable intestinal first-pass effect of DA-8159 in rats
(18).
In the rat model of
dehydration, hematocrit was significantly greater than controls after both intravenous
and oral administration of DA-8159 (Tables 2 and 4). Similar results were also
reported from other rat studies (7,11,28–30). The
binding of DA-8159 to blood cells was considerable; the mean plasma-to-blood
cells concentration ratios of DA-8159 in three rabbit blood at initial DA-8159
blood concentrations of 1–10 mg/ml were 0.662–0.812 (25). The bound
fractions of adriamycin (31) and propranolol (32) to
red blood cells were reported to act as barriers for elimination. Hence, the
significantly greater hematocrit value in rat model of dehydration (Table 2 and
4) could influence at least partly to the slower CL of DA-8159 in rat model of
dehydration (Table 2).
CONCLUSIONS
After intravenous administration of DA-8159 in rat model of dehydration,
the CLNR of DA-8159 was comparable to controls (Table 2), since
DA-8159 was metabolized mainly via CYP3A1/2 (5) and CYP3A1/2 was not changed in
rat model of dehydration (12). However, the CL of DA-8159 was significantly slower than
controls, and this could be at least partly due to significantly slower CLR
of DA-8159 in rat model of dehydration (Table 2). The slower CLR
could be due to significantly smaller Ae0–24 h,
DA-8159 in rat model of dehydration (Table 2), and this could be due to
urine flow rate-dependent CLR of DA-8159 in rats (26) and impaired
kidney function in rat model of dehydration. After intravenous administration
of DA-8159 in rat model of dehydration, the significantly greater AUC of
DA-8164 was possibly due to significantly greater exposure of the parent drug (the
significantly greater AUC of DA-8159), and significantly greater AUC of DA-8164
in rat model of dehydration (Table 3). After oral administration of DA-8159,
the AUC values of both DA-8159 and DA-8164 were comparable between two groups
of rats and this could be due to changes in intestinal first-pass effects in rat
model of dehydration.
ACKNOWLEDGMENTS
This study was supported in part by a grant from the Korea Ministry of
Health and Welfare, 02-PJ2-PG4-PT01-0024.
Nonstandard abbreviations:
HPLC, high-performance liquid chromatography;
Vmax, maximum velocity; Km, Michaelis–Menten
constant; CLint, intrinsic
clearance; AUC, total area under
the plasma concentration–time curve from time zero to time infinity; MRT, mean residence time; Vdss, apparent volume of
distribution at steady state; CL, time-averaged
total body clearance; CLR, time-averaged
renal clearance; CLNR, time-averaged
nonrenal clearance; Ae0–24 h, total
amount excreted in 24-h urine; GI24
h, total amount recovered from the entire
gastrointestinal tract (including its contents and feces) at 24 h; Cmax, maximum plasma
concentration; Tmax, time
to reach a Cmax; F, extent of absolute oral
bioavailability.
REFERENCES
[1]
Shim,
H.J., Kim, Y.C., Lee, J.H., Kwon, J.W., Kim, W.B., Kim, Y.G., Kim, S.H., and Lee,
M.G.., Interspecies pharmacokinetic scaling of DA-8159, a new erectogenic, in
mice, rats, rabbits, and dogs, and prediction of human pharmacokinetics. Biopharm Drug Dispos, 26:269–277, 2005.
[2] Doh,
H., Shin, C.Y., Son, M., Ko, J.I., Yoo, M., Kim, S.H.,
and Kim, W.B., Mechanism of erectogenic effect of the selective
phosphodiesterase type 5 inhibitor, DA-8159. Arch Pharm Res, 25:873–878, 2002.
[3] Ahn,
B.O., Kang, K.K., Ahn, G.J., Kwon, J.W., Kim, W.B., Kang, K.S., and Lee, Y.S.,
Efficacy of DA-8159, a new PDE5 inhibitor, for inducing penile erection in
rabbits with acute spinal cord injury. Int
J Impot Res, 15:405–411, 2003.
[4] Kang,
K.K., Ahn, G.J., Ahn, B.O., Yoo, M., and Kim, W.B., DA-8159, a new PDE5
inhibitor, induces penile erection in conscious and acute spinal cord injured
rabbits. Eur Urol, 43:689–695, 2003.
[5] Kim,
Y.C., Shim, H.J., Lee, J.H., Kim, S.H., Kwon, J.W., Kim, W.B., and Lee, M.G..,
Effect of enzyme inducers and inhibitors on the pharmacokinetics of intravenous
DA-8159, a new erectogenic, in rats. Biopharm Drug Dispos, 26:233-241, 2005.
[6] Bakar,
S.K., and Niazi, S., Effect of water deprivation on aspirin disposition
kinetics. J Pharm Sci, 72:1030-1034, 1983.
[7] Huang,
J.Y., Kim, O.N., Lee, S.H., and Lee, M.G., Effects of water deprivation on the
pharmacokinetics and pharmacodynamics of bumetanide in rats. Biopharm Drug Dispos, 14:463-474,
1993.
[8] Ha,
H.A., Lee, S.H., Kim, S.H., Kim, O.N., and Lee, M.G., Effect of water
deprivation for 48 hours on the pharmacokinetics and pharmacodynamics of azosemide
in rats. Res Commun Mol Pathol Pharmacol,
93:109-128, 1996.
[9] Son,
I.J., Moon, Y.J., Lee, M.G., and Sohn
YT. No effect of water
deprivation for 48 hours on the pharmacokinetics of intravenous tacrolimus in
rats. Res Commun Mol Pathol Pharmacol,
107:279-289, 2000.
[10] Kim,
S.H., Kwon, J.W., Kim, W.B., Lee, I., and Lee,
M.G. Effects of water deprivation for 72 hours on the pharmacokinetics of a new
carbapenem, DA-1131, in rats. Life Sci,
71:2291–2298, 2002.
[11] Kim,
Y.C., Kim, Y.G., Kim, E.J., Cho, M.K., Kim, S.G., and Lee MG., Pharmacokinetics
of intravenous chlorzoxazone in rats with dehydration and rehydration: Effects
of food intakes. Biopharm Drug Dispos,
24:53–61, 2003.
[12] Kim,
S.G., Kim, E.J., Kim Y.G., and Lee, M.G., Expression of cytochrome P-450s and
glutathione S-transferases in the rat liver during water deprivation: Effects
of glucose supplementation. J Appl
Toxicol, 21:123-129,
2001.
[13] Bae,
S.K., Lee, S.J., Kim, J.W., Kim, Y.H., Kim, S.G., and Lee, M.G.,
Pharmacokinetics of oltipraz after intravenous and oral administration in rats
with dehydration for 72 hours. Biopharm Drug Dispos, 26:77-83, 2005.
[14] Bradford,
M.M., A rapid and sensitive method for the quantitation of microgram quantities
of protein utilizing the principle of protein–dye binding. Anal Biochem, 72:248–254, 1976.
[15] Shim,
H.J., Lee, E.J., Jung, Y.H., Kim, S.H., Kim, S.H., Yoo, M., Kwon, J.W., Kim, W.B.,
and Lee, M.G., Determination of a new phosphodiesterase V inhibitor, DA-8159,
in plasma and urine by high-performance liquid chromatography. J Pharm Biomed Anal, 30:527–533, 2002.
[16] Lineweaver,
H., and Burk, D., The determination of enzyme dissociation constants. J Am Chem Soc, 56:658–666, 1934.
[17] Kim,
S.H., Choi, Y.M., and Lee, M.G., Pharmacokinetics and pharmacodynamics of
furosemide in protein–calorie malnutrition. J
Pharmacokinet Biopharm, 21:1-17, 1993.
[18] Shim,
H.J., Kim, Y.C., Park, K.J., Kim, D.S., Kwon, J.W., Kim, W.B., and Lee, M.G.,
Pharmacokinetics of DA-8159, a new erectogenic, after intravenous and oral
administration to rats: Hepatic and intestinal first-pass effects. J Pharm Sci, 92:2185–2195, 2003.
[19] Shim,
H.J., Lee, E.J., Kim, S.H., Kim, S.H., Yoo, M., Kwon, J.W., Kim, W.B., and Lee,
M.G., Factors influencing the protein binding of a new phosphodiesterase V
inhibitor, DA-8159, using an equilibrium dialysis technique. Biopharm Drug Dispos, 21:285–291, 2000.
[20] Chiou,
W.L., Critical evaluation of potential error in pharmacokinetic studies using
the linear trapezoidal rule method for the calculation of the area under the
plasma level–time curve. J Pharmacokinet Biopharm,
6:539-546, 1978.
[21] Gibaldi,
M., and Perrier, D., Pharmacokinetics. 2nd ed. Marcel–Decker, New York, NY,
USA,
1982.
[22] Chiou,
W.L., New calculation method for mean apparent drug volume of distribution and
application to rational dosage regimens. J
Pharm Sci, 68:1067-1069,
1979.
[23] Eatman,
F.B., Colburn, W.A., Boxenbaum, H.G., Posmanter, H.N., Weinfeld, R.E., Ronfeld,
R., Weissman, L., Moore, J.D., Gibaldi, M., and Kaplan, S.A.,
Pharmacokinetics of diazepam following multiple-dose oral administration to
healthy human subjects. J Pharmacokinet Biopharm,
5:481-494, 1977.
[24] Chiou,
W.L., New calculation method of mean total body clearance of drugs and its
application to dosage regimens. J Pharm
Sci, 69:90-91, 1980.
[25] Shim,
H.J., Lee, E.J., Kim, S.H., Kim, S.H., Yoo, M., Kwon, J.W., Kim, W.B., Lee, H.S.,
and Lee, M.G., Pharmacokinetics, stability, and blood partition of DA-8159, a
new phosphodiesterase V inhibitor. Res
Commun Mol Pathol Pharmacol, 108:275–286, 2000.
[26] Kim,
Y.C., Kwon, J.W., Kim, W.B., Lee, I., and Lee, M.G., Pharmacokinetic changes of
intravenous DA-8159, a new erectogenic, in rat with diabetes mellitus induced
by streptozotocin. J Pharm Sci, 93:2374–2387,
2004.
[27] Lee,
M.G., and Chiou, W.L., Evaluation of potential causes for the incomplete
bioavailability of furosemide: Gastric first-pass metabolism. J Pharmacokinet Biopharm, 11:623–640,
1983.
[28] Kutscher,
C.L., Hematocrit, plasma osmolality, and plasma protein concentration as
estimators of plasma volume in hooded rats during food and water deprivation. Physiol Behav, 7:283–285, 1971.
[29] LeCompte,
J., Dumont, L., Hill, J., du Souich, P., and Lelorier,
J., Effect of water deprivation and rehydration on gentamicin disposition in
the rat. J Pharmacol Exp Ther, 218:231–236,
1981.
[30] Hope,
A., and Tyssebotn, I., The effect of water deprivation
on local renal blood flow and filtration in the laboratory rat. Circ Shock, 11:175–186, 1983.
[31] Lee,
H.J., and Chiou, W.L., Erythrocytes as barriers for drug elimination in the
isolated rat liver. I. Doxorubicin. Pharm Res, 6:833-839, 1989.
[32] Lee,
H.J., and Chiou, W.L., Erythrocytes as barriers for drug elimination in the
isolated rat liver. II. Propranolol. Pharm Res, 6:840-843, 1989.
JPPS Contents
Published by the Canadian Society for
Pharmaceutical Sciences.
Copyright © 1998 by the Canadian
Society for Pharmaceutical Sciences.
http://www.cspscanada.org/
CSPS Home |
JPPS Home |
Search |
Subscribe to JPPS