J Pharm Pharmaceut Sci (www.cspscanada.org) 9(1):119-123, 2006
The role of nitric oxide and protein kinase C in lipopolysaccharide-mediated vascular hyporeactivity
Gholamreza Karimi1,
Zahra Fatehi2, Zahra Gholamnejad2
1-Department of Pharmacodynamy and Toxicology. Pharmacy School, Mashhad, Iran
2-Department of Physiology, Medical School, Mashhad, Iran
Received 20 January 2006, Revised 22 February 2006, Accepted 23 February 2006, Published 09 March 2006
Corresponding Author: Gholamreza Karimi, Department of Pharmacodynamy and Toxicology. Pharmacy School, Mashhad, Iran. gho_karimi@yahoo.com
Abstract PURPOSE: Overactivation of nitric oxide and protein kinase C (PKC) pathway has been reported to play a role in the pathogenesis of vascular hyporesponsiveness of endotoxic shock. In this study we investigated the role of nitric oxide and PKC in lipopolysaccharide (LPS) mediated vascular hyporeactivity. METHODS: Contraction to phenylephrine and endothelium-dependent and independent vasodilation in the presence and absence of a nonspecific NO inhibitor (L-NAME) and potent PKC inhibitor (chelerythrine) were examined. RESULTS: In LPS treated rats, contractile response of aortic rings to phenylephrine and relaxation in response to acetylcholine were reduced, but relaxation induced by sodium nitroprusside remained unchanged. The attenuation of contractile response to phenylephrine in the presence of L-NAME and chelerythrine was more pronounced in aortic ring isolated from LPS treated rats than control. L-NAME decreased acetylcholine -dependent vasodilation in both group but it was more pronounced in LPS treated rats. Chelerythrine pretreatment improved maximal relaxation to acetylcholine in aortic ring isolated from LPS treated rats. CONCLUSION: These data indicate that the vascular hyporesponsiveness to phenylephrine and acetylcholine after treatment with LPS may be related to an enhanced NO production in the smooth muscle cells and PKC plays a role as an intracellular mediator of LPS-induce NOS activity and vascular suppression.
INTRODUCTION
Septic shock is the most common cause of death in medical and surgical intensive care units (1-2). In humans, it is characterized by a high cardiac output and decreased systemic peripheral resistance due to dilation of resistance arteries (3-4). This cause progressive systemic hypotension that is resistant to vasoconstrictors (5-6) and lead to abnormal organ perfusion and so organ damage and failure (7- 9).
The most Common cause of septic shock is infection with gram-negative bacteria, resulting in the release of bacterial lipopolysaccharid (LPS) (10-11). Many mediators which markedly influence cardiovascular function are generated in response to LPS. These vasoactive substances include catecholeamines, histamine, thromboxanes, leukoterines, angiotensines, various as yet uncharacterized 'shock toxins' and oxygen derived free radicals (12-13). Nitric oxide (NO) is perhaps the latest mediator to be scrutinized with reference to the pathophysiology of sepsis (5, 14). LPS is known to express an inducible isoform of NOS (iNOS), followed by production of large amount of NO in various cells, including vascular smooth muscle, endothelial cells and macrophage, which contributes importantly to several key feature in pathophysiology of septic shock, such as hypotension and vascular hyporeactivity to vasoconstrictor agent (12,15) and vasodilators (16-17).
Protein kinase C plays a role as intracellular mediator of LPS-induced NOS activity and vascular suppression (18-20). Lipopolysaccharid induce time-dependent increase in PKC isotype mRNA expression and isotype-specific PKC activation and synthesis in vascular tissues (21). It has shown that PKC plays an important role in LPS-induced NO formation (22-23).
In this study, we investigated the effect of NO pathway, by blocking NO synthase with L-NAME and of PKC pathway, by blocking with chelerythrine, on the responsiveness of the isolated aortic rings of LPS-treated rats to vasoconstrictor agent (Phenylephrine) and an endothelium-dependent vasodilator (Acetylcholine) and an endothelium-independent vasodilator sodium nitroprusside.
MATERIALS
AND METHODS
Animals
Adult male Sprague Dawley rats, (Razi Institutes, Mashhad, Iran) weighing between 250-300 g were used. Animals were housed in temperature and humidity controlled, light-cycled quarters. The protocols used conformed to guidelines of the conduct approved by the committee on the ethics of animal experiments in Mashhad University. Animals were randomly divided into two groups including control and LPS treated. Control rats received saline injection (1ml kg-1 i.p. n=5), whereas septic rats received a bolus injection of LPS (10 mg kg-1 i.p. n=5) 5 hours before examination.
Preparation of rat thoracic aortic rings
Five hours after saline or LPS injection, animals were anesthetized with sodium pentobarbital (60 mg kg-1 i.p.) and thoracic aorta was immediately removed and placed in physiologic salt solution (containing (in mM) : NaCl,130; KCl, 4.7; CaCl2, 1.6; MgSO4, 1.17; KH2PO4, 1.18; NaHCO3, 14.9; Dextrose, 5.5; and EDTA-Ca,Na2, 0.03) bubbled with mixture of 95% O2 and 5% CO2. After cleaning the tissue of fat and other adhering tissues, the vessel were cut into 3 mm long rings, with special care to avoid damaging the endothelium. The preparation were mounted on a pair of stainless-steel hooks; one of each was fixed to a L-shaped rod inside the chamber and other to an isometric force transducer (F-60, Narco Biosystems, Inc., TX, USA) connected to a polygraph (MK III-S, Narco Biosystems, Inc., TX, USA). Tissues were allowed to equilibrate under an optimum final force of 1.5 g for a period of 60 min in a water-jacketed tissue bath (10 ml) containing oxygenated physiologic salt solution at 37°C (final pH of 7.4), renewing the buffer every 15 min. After stabilization vascular smooth muscle integrity was assessed by standard contraction obtained with 40 mM KCl. Endothelium integrity was assessed with 1μM acetylcholine.
Experimental procedure
All of the following experiments were conducted on the aortic rings of both LPS-treated and control rats.
First. Concentration response was assessed by adding cumulative concentration of phenylephrine (0.1nM to 1μM). The concentration-contraction curves were studied for: (1) intact aortic rings; (2) aortic rings incubated with L-NAME (10μM) for 20min; (3) aortic rings incubated with chelerythrine (10μM) for 20min.
Second. Relaxation response to acetylcholine, an endothelium-dependent vasorelaxant, was assessed by adding cumulative concentration of acetylcholine (1nM to1mM) on the aortic rings precontracted with phenylephrine 1 μM. The similar concentration-relaxation curves were also done on: (1) the aortic rings incubated with L-NAME (10μM for 20min); (2) aortic rings incubated with chelerythrine (10μM for 20min). Relaxation response curves to sodium nitroprusside (SNP) were also constructed by adding cumulative concentration (1nM to 10nM) to phenylephrine- precontracted aortic rings.
Statistical Analysis of Data
Results are expressed throughout as means ± S.E.M. and were analyzed by one way analysis of variance (ANOVA) followed by a Tukey-Kramer multiple comparison test (for comparison of responses to phenylephrine and acetylcholine in aortic). P value of less than 0.05 was considered to be significant.
RESULTS
Phenylephrine-induced contraction responses
Phenylephrine produced a concentration-dependent contraction in aortic rings. Contractile responses to phenylephrine were significantly decreased in aortic rings isolated from LPS treated rats (Figure 1).
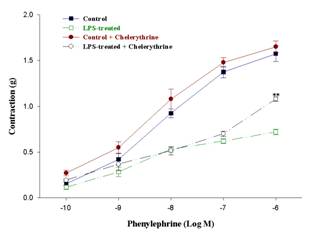
Fig.1. Dose-response curves of the contraction of aortic
rings of the control and LPS treated rats to phenylephrine. It also shows the
effect of incubation with chelerythrine (10μM for 20min). (n = 5, mean ±
SEM, (**) P< 0.01 compared to the value of LPS-treated).
Inhibition of PKC by chelerythrine had no significant effect on the maximal response (Rmax) of aortic rings of the control rats. However, in the aortic rings of LPS treated groups, chelerythrine incubation caused significant increase on Rmax compared to respective intact rings (Figure 1).
Preincubation with L-NAME significantly increased responses to phenylephrine in the aortic rings of LPS treated rats compared to control (Figure 2).
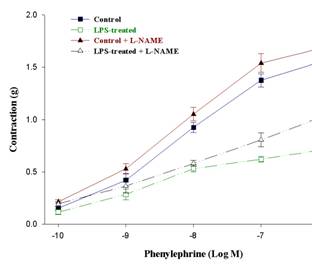
Fig.2.
Dose-response curves of the
contraction of aortic rings of the control and LPS treated rats to
phenylephrine. It also shows the effect of incubation with L-NAME (10μM
for 20min). (n = 5, mean ± SEM, (*) P< 0.05 compared to the value of
LPS-treated).
Acetylcholine -induced relaxation responses
Acetylcholine caused a concentration-dependent relaxation in the precontracted aortic rings. In the aortic rings of LPS treated rats the relaxation responses were significantly less than control groups (figure 3).
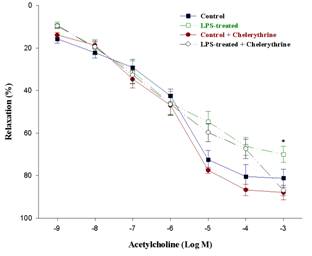
Fig.3. Acetylcholine-induced relaxation response curves of
aortic rings of the control and LPS treated rats. It also shows the effect of
incubation with chelerythrine (10μM for 20min). (n = 5, mean ± SEM, (*)
P< 0.05 compared to the value of LPS-treated + chelerythrine).
Preincubation with chelerythrine increased the maximal relaxation response of the aortic rings of LPS treated animals. However, there was no significant change in the concentration-response curve of acetylcholine in control groups (figure 3). Incubation with L-NAME caused significant attenuation of acetylcholine induced relaxation of aortic rings in both groups (figure 4).
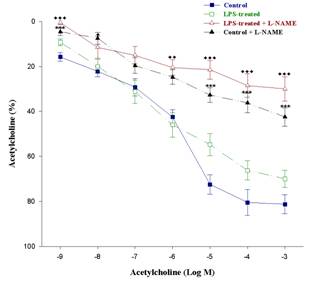
Fig.4. Acetylcholine-induced relaxation response curves of
aortic rings of the control and LPS treated rats. It also shows the effect of
incubation with L-NAME (10μM for 20min). (n = 5, mean ± SEM, (♦)
P< 0.05, (♦♦) P< 0.01, (♦♦♦) P< 0.001
compared to the value of LPS-treated and (***) P< 0.001 compared to the
value of control.
Sodium nitroprusside-induced relaxation responses
Sodium nitroprusside induced a concentration-dependent relaxation in the precontracted aortic rings (data not showed). There was no significant difference in sodium nitroprusside-induced relaxation of the aortic rings between the LPS treated and control rats.
DISCUSSION
The data presented here show that LPS can induce vascular hyporesponsiveness to vasoactive factors. Different hypothesis for the vasodilation and resistance to vasopressors that occur in this syndrome had been pronounced such as activation of potassium channels (KATP and KCa channels) in the plasma membrane of vascular smooth muscle and activation of the inducible form of nitric oxide synthase (24-25).
In our experiments the pressor response to phenyleprine was impaired in LPS-treated rats and blocking NO pathway improved it. This may be due to activation of the inducible form of nitric oxide synthase in aorta of LPS treated rats.
Our data also showed that LPS reduced endothelium-dependent vascular reactivity to receptor-mediated agonists such as acetylcholine. The effect of LPS on the aortic NO-mediated component of acetylcholine-induced relaxation was not caused by a reduction in the sensitivity of vascular smooth muscle to NO, because responses to SNP were unaltered. This finding confirm the theory that by the time iNOS is up regulated in endotoxic shock, there is a corresponding down-regulation in the endothelial constitutive NOS (eNOS) (26-28).
Excessive iNOS activation and NO production is not the main cause of these defects and other factors like PKC directly or indirectly may contribute to vascular hypocontractility. In our study chelerythrine showed beneficial effect on maximal contractile and dilatory response of LPS-treated rat which can suggest the probable effect of PKC in reducing response of aorta to phenylephrine and acetylcholine. This is in agreement with previous studies which showed that LPS activates PKC and induce iNOS transcription and NO formation (20, 21, 29). The relationship between LPS stimulation, PKC activation, iNOS expression and NO formation are complex, and it is likely that PKC influence this process at several sites in the cell. Those observations suggest that multiple intracellular pathways, some of which are PKC independent, can transduce an LPS signal into iNOS expression and PKC activity is necessary for NO formation after iNOS induction. It can be concluded that PKC and NO pathways are altered in aortic rings of LPS treated rats. The exact nature of the altered vascular responses needs further detailed studies.
REFERENCES
(1) Astiz ME, and Rackow EC. Septic
shock. Lancet, 351: 1501-1505, 1998.
(2) Glauster MP.
Pathophysiologic basis of sepsis: considerations for future strategies of
intervention. Crit Care Med, 28: S4-S8, 2000.
(3) Groeneveld ABJ, Bronsveld
W, Thijs LG. Hemodynamic determinants of mortality in human septic shock.
Surgery, 99: 140-152, 1986.
(4) Landry DW, and Oliver JA. The pathogenesis of
vasodilatory shock. N Engl J Med, 345: 588-595, 2001.
(5) Macarthur
H, Westfall TC, Riley DP, Misko TP, Salvemini D. Inactivation of catecholamines
by superoxide gives new insights on the pathogenesis of septic shock. Proc Natl
Acad Sci, 97: 9753–9758, 2000.
(6) Temple GE, Brown AN, Morinelli TA, Halushaka PV, Cook JA,
Changes in vascular responsiveness to thromboxane mimetic in endotoxic
tolerance. Shock, 16:389-392, 2001.
(7) Lang CH, Bagby GJ, Ferguson JL, Spitzer JJ. Cardiac output and
redistribution of organ blood flow in hypermetabolic sepsis. Am J Physiol, 246:
R331-R337, 1984.
(8) Mulder MF, Van
Lambalgen AA, Van den Bos GC, Thijs LG. The
fall of cardiac output in endotoxemic rats can not explain all changes in organ
blood flow: a comparison between endotoxin and low venous return shock. Shock,
5:135-140, 1996.
(9) Piepot HA, Boer C, Groenveled ABJ, Sipkema P.
Lipopolysaccharide impairs endothelial nitric oxide synthesis in rat renal
arteries. Kidney Int, 57:2502-2510, 2000.
(10) Abraham E, Wunderink R,
Silverman H, et al. Efficacy
and safety of monoclonal antibody to human tumor necrosis factor a in patients with sepsis syndrome. JAMA, 273: 934–941, 1995.
(11) Marsh CB, Wewers MD. The pathogenesis of sepsis: factors that
modulate the response to gram-negative bacterial infection. Clin Chest Med, 17:
183-197, 1996.
(12) Parratt JR. Nitric oxide in sepsis and
endotoxemia. J Antimicrob
Chemother, 41: 31-39, 1998.
(13) Stoclet JC, Martinez MC, Ohlamann P, Chasserot S, Schott C,
Kleschyov AL, Schneider F. and Andriantsitohaina R. Induction of nitric oxide
synthase and dual effects of nitric oxide and cyclooxygenase products in
regulation of arterial contraction in human septic shock. Circulation, 100:
107-112, 1999.
(14) Miyamoto A, Yamazaki Y, Takagi T, Ishiguro S, Nishio A.
Enhancement of endotoxin-induced vascular hyporeactivity to phenylephrine in
the thoracic aortas of Mg-deficient rats ex vivo. Life Sci, 73: 2713-2726,
2003.
(15) Farmer MR, Roberts
RE, Gardiner SM, Ralevic V. Effects of in vivo lipopolysaccharide infusion on
vasoconstrictor function of rat isolated mesentery, kidney and aorta. J
Pharmacol Exp Ther, 306: 538-545, 2003.
(16) Farias NC, Borelli-Montigny GL, Fauaz G, Feres T, Borges AC.
Different mechanism of LPS-induced vasodilation in resistance and conductance
arteries from SHR and normotensive rats. Br J Pharmacol, 137: 213-220, 2002.
(17) Piepot
HA, Groeneveld ABJ, Van Lambalgen
AA, Sipkema P. Endotoxin impairs
endothelium-dependent vasodilation more in the coronary and renal arteries than
in other arteries of the Rat. J Surg Res, 110: 413-418, 2003.
(18) Lidington D, Ouellette Y, Tyml K. Endotoxin increases
intracellular resistance in microvascular endothelial cells by a tyrosine
kinase pathway. J Cell physiol, 185:117-125, 2000.
(19) Han YL, Kang J, Li SH. Protein kinase C and protein tyrosine
kinase mediate lipopolysaccharide- and cytokine-induced nitric oxide formation
in vascular smooth muscle cells of rats. Sheng Li Xue Bao, 55:265-272, 2003.
(20) McKenna
TM, Clegg JM, Williams TJ. Protein kinase C is a mediator of
lipopolysaccharide-induced vascular suppression in the rat aorta.
Shock, 2: 84-89, 1994.
(21) McKenna
TM, Fan SX, Li S. Lipopolysaccharide-responsive protein
kinase C isotypes in the adult rat aorta. Shock, 7:269-273, 1997.
(22) Tremblay P,
Houde M, Arbour N, Rochefort D, Masure S, Opdenakker G, Oth D. Differential
effects of PKC inhibitors on gelatinase b and interleukin 6 production in the
mouse macrophage. Cytokine, 7:130-136, 1995.
(23) Li S, Huang FL, Feng Q, Liu J, Fan SX, McKenna TM. Overexpression
of protein kinase C alpha enhances lipopolysaccharide-induced nitric oxide
formation in vascular smooth muscle cells. J Cell Physiol, 176:402-411, 1998.
(24) Pleiner J,
Heere-Ress E, Langenberger H, Sieder AE, Bayerle-Eder M, Mittermayer F,
Fuchsjager-Mayrl G, Bohm J, Jansen B, Wolzt M.
Adrenoceptor hyporeactivity is responsible for Escherichia Coli
endotoxin-induced acute vascular dysfunction in humans. Arterioscler Thromb
Vasc Biol, 22:95-100, 2002.
(25) Wu MS, Yang CW, Bens M, Yu HM, Huang JY, Wu CH, Huang CC,
,Vandewalle A. Cyclosporin inhibits nitric oxide production in medullary
ascending limb cultured cells. Nephrol Dial Transplant,
13:2814-20, 1998.
(26) Yoshizumi M, Perrella MA, Burnett JC,
Lee ME. Tumor necrosis factor downregulates an endothelial nitric
oxide synthase mRNA by shortening its half-life. Circ Res,
73:205-209,1993.
(27) Liu SF, Adcock
IM, Old RW, Barnes PJ, Evans TW. Differential regulation of
the constitutive and inducible nitric oxide synthase mRNA by lipopolysaccharide
treatment invivo in the rat. Crit Care Med, 24: 1219-1225, 1996.
(28) Gunnett C A, Lund
DD, Chu Y, Brooks II RM, Faraci FM, Heistad DD. NO-dependent vasorelaxation is
impaired after gene transfer of inducible NO-synthase. Arterioscler Thromb Vasc
Biol, 21:1281-1287, 2001.
(29) Paul A, Doherty K, Plevin R. Differential regulation by protein kinase C isoforms of nitric oxide synthase induction in RAW 264.7 macrophages and rat aortic smooth muscle cells. Br J Pharmacol, 120: 940-946, 1997.
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
CSPS Home | JPPS Home | Search | Subscribe to JPPS