J Pharm Pharmaceut Sci (www.cspscanada.org) 8(3):409-418, 2005
Development of cyclodextrin microspheres
for pulmonary drug delivery
Malika Skiba, Frédéric Bounoure, Cécile Barbot,
Philippe Arnaud, Mohamed Skiba
Laboratoire de Pharmacie
Galénique et Biopharmacie, ADEN-UPRES EA
3234, UFR de Medecine-Pharmacie, Rouen Cedex, France
Received March 1, 2005 ,Revised June 21, 2005, Accepted June 28, 2005, Published August 24, 2005
Corresponding author. Mohamed Skiba Laboratoire de Pharmacie Galénique et Biopharmacie, ADEN-UPRES EA 3234, UFR de
Medecine-Pharmacie, 22 Boulevard Gambetta, 76183 Rouen Cedex, France.
ABSTRACT
Purpose. Microparticles of
diameter < 5 mm were synthesized
by interfacial cross-linking of 7.5% (w/v) b-cyclodextrins (b-CD) with 4.5%
(w/v) terephtaloyle chloride in 1 M NaOH, in order to provide stable vector for
drug encapsulation suitable for administration at the alveolar scale. Methods. Batches were prepared varying
different parameters such as amount of monomer (b-CD) (5-30% w/v), NaOH
concentration (0.5-4 M), reaction time (15-240 min), agitation rate (8000-24000
rpm), amount of cross-linking agent (terephtaloyle chloride: 1.25-10% w/v),
surfactant percentage (2.5-10% of Span 85), studying the influence of the
freeze-drying step. Microparticles were controlled with respect to their size
by a laser diffraction technique, pH of the colloidal suspension, IR
spectroscopy, Differential Scanning Calorimetry. After optimization of the
microparticles size, complexation with amikacin sulfate was investigated
comparing encapsulation efficiency and yield at each step of the preparation
(solubilization, emulsification, cross-linking, freeze-drying), contact time
and influence of the amount of amikacin. Results.
An optimized method was obtained with 1 M NaOH, 4.5% (w/v) cross-linking agent
and 5% (w/v) surfactant agent, a 30 min reaction time, a 24000 rpm agitation
rate, conducting to microparticles whose size is inferior to 5 mm. Amikacin sulfate
encapsulation in polycondensed b-cyclodextrin showed
that better incorporation was obtained during the solubilization step or just
before freeze-drying. Conclusions.
Amikacin encapsulation in 5 mm diameter
microparticles of b-CD is achievable
for pulmonary drug delivery.
INTRODUCTION
Native cyclodextrins are
polysaccharides made up of six to eight cyclic linked oligosaccharides of
D-glucopyranose monomers connected by a-1, 4-indican
bonds. These compounds form cone-shaped molecules with primary hydroxyl groups
(6-OH) arranged in an inner hydrophobic cavity of 5.7, 7.8 and 9.5 Å
respectively for a-, b-, g-cyclodextrins, and
secondary hydroxyl groups (2- and 3-OH) rendering external walls hydrophilic
(1). These two microenvironments confer to the molecule the ability of forming
inclusion complex with guest molecules. Cyclodextrins act as molecular hosts
toward various, poorly water-soluble drugs, ranging from ion, very polar
molecules to non-polar molecules, affecting advantageously their
physicochemical properties (2, 3). Thus, they have found extensive applications
in chromatography, catalysis, asymmetric reactions, food, cosmetic,
pharmaceutical technology (4), medicinal applications. Partially or entirely
encapsulation occurs by the intermediate of hydrophobic forces and van der
Waals interactions, ion pairing, hydrogen bonding (5) participating in
improving, through complexation, the aqueous solubility and stability of drugs
(6-8), vitamins and food colorant, preventing molecules self-aggregation,
ameliorating dissolution rate, bioavailability of the hydrophobic drugs,
decreasing toxicity and controlling drug releasing (9-11).
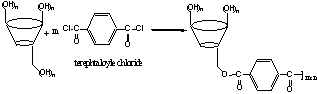
Scheme 1. Preparation of the CD microparticles with
terephtaloyle chloride.
Preparation of
inclusion complexes (4, 12, 13) can be done by different methods namely,
coprecipitation, freeze-drying, kneading, grinding or co-pulverizing, microwave
heating (14). Inclusion complexes can be formed in solution or in a solid state
(15). The advantage of the preparation in the crystalline state is the
protection of the complexes formed against some type of reactions such as
oxidation, hydrolysis and the role played in the decrease of their sublimation
and volatility (16). Guests of varying size were tested by many authors,
screening their physicochemical features in function of various stoechiometric
ratios (17, 18).
Analytical methods
for physicochemical characterization used are fluorescence spectroscopy (steady
state fluorescence), NMR spectroscopy, IR (19), differential scanning
calorimetry (DSC), elemental analysis, power X-ray diffraction, thermogravimetric
analysis (TGA). When complexed with CDs, modification in the photoreactivity
(20, 21) of the guest molecule occurs and generally fluorescence efficiencies
are enhanced (22, 23) due to a decrease in non-radiative and quenching
processes observed in bulk solution (24, 25). Fluorimetric analysis is also
used due to its sensibility and selectivity to evaluate association constants
of complexes (26-28). NMR gives useful informations about the geometry of
complexes (29-31). Therefore, cyclodextrin microspheres have been shown to be
stable vectors for drug encapsulation (10, 30) and may have some applications
in pulmonary drug delivery when deposited in the alveolar region.
Two methods for the
preparation of drug-containing CD particles are described in the litterature.
The first one involves cross-linking drug directly to CDs adsorbed on a porous
inorganic oxide via a bifunctional agent, epichlorydrin (32) or sebacoyle
chloride (33). The second one requires the synthesis of 10-35 mm microcapsules, in an
emulsion system, and subsequent interfacial cross-linking of b-cyclodextrin (b-CD) with
terephtaloyle chloride (34). In this paper, we describe our efforts to optimize
the second method to produce 5 mm particles intended for
alveolar drug delivery following inhalation. Some authors prepared
microparticles, with a mean geometric size inferior to 5 mm, from an oil-in-water
emulsion consisting of an aqueous phase containing cyclodextrin derivative
(35). The first goal of our investigation was devoted to the preparation of
microparticles from native cyclodextrins synthesized by interfacial
cross-linking with terephtaloyle chloride. We measured the influence of
reaction conditions on the resulting microparticle size. Once our objectives
were reached, fabrication yield and pH of the colloidal suspension (nearest
physiological pH) were optimized and microparticles characterized by
granulometry, FT-IR and DSC. The second part of the study concerned the
microparticle complexing properties with amikacin sulfate (Figure 1), a
potential drug to be administered in pulmonary disease. The effects of
variations in the preparation conditions of encapsulated microparticles were
investigated. Amikacin is an antibiotic of the class of aminosides used in the
treatment of severe infections, particularly those due to aerobic,
Gram-negative bacilli (GNB). Their main drawback has been the occurrence of
(reversible) nephrotoxicity and ototoxicity in a significant number of patients
Nosocomial pneumonia with GNB is the first cause of infection mortality for
patients requiring mechanical ventilation. The pulmonary targeting through
encapsulation of this antibiotic into cyclodextrin microspheres seems to be
interesting to optimize therapeutic efficacy and limit it’s toxicity. Particular
system based on cyclodextrin microspheres presents a great stability, a high
encapsulation efficiency and allows drug spray-drying at the alveolar scale as
compared with other drug delivery systems.
Figure 1. Amikacin sulfate.
Microspheres were
synthesized by interfacial cross-linking of b-cyclodextrins (b-CD) with
terephtaloyle chloride in 1 M NaOH (Scheme 1). A series of batches were
prepared in which formulation parameters such as monomer concentration (5-30%
w/v), NaOH concentration (0.5-4M), reaction time (15-240min), agitation rate
(8,000-24,000 rpm), polymerization agent concentration (1.25-10% w/v), and
surfactant content (2.5-10% of Span 85) were varied. The influence of
freeze-drying on the particles size was also investigated. Microspheres size
and pH of the colloidal suspension were optimized around two objectives. (1)
The maximum yield of particles smaller than 5mm, measured by a laser
diffraction technique, and (2) pH nearest to physiological pH. Following size
and pH optimization, amikacin sulfate was encapsulated into the microspheres at
four different stages of the fabrication process (solubilization,
emulsification, cross-linking and freeze-drying) and the encapsulation
efficiency (mass of amikacin sulfate in particles (mg)/ mass of b-CD (mg)) and yield
(mass of amikacin sulfate in particles (mg)*100/mass of amikacin
sulfate introduced (mg)) were determined
by a HPLC method (36).
MATERIAL
AND METHODS
Material
Amikacin sulfate was purchased
from Bristol-Myers Squibb (France). b-cyclodextrin was
obtained from Roquette Frères (
Preparation
of microparticles
Polycondensation of b-cyclodextrin by
terephtaloyle chloride was undertaken by the following standard method :
In a first step, 6
mL of b-CD (7.5% w/v) was
solubilized in 1M NaOH and emulsified during 10 min, using a Heidolph RGL 500
stirring motor (Prolabo, France) at a stirring rate of 2000 rpm, in a 30
mL-cyclohexane solution containing 5% (v/v) sorbitan 85 trioleate, at ambiant
temperature.-
-Cross-linking solution was
prepared dissolving 5% (w/v) terephtaloyle chloride in a (1:4, v/v) mixture
chloroform/cyclohexane.
- Microparticles were formed by addition of this organic
phase to the emulsion, and mixing with 30 min stirring, the agitation speed
being regulated to 2000 rpm. In this study, on contrary to the standard method,
agitation rate was regulated to 8000 rpm (Ultra Turrax type TP 18/10 Janke et
Kunkel). This cross-linking reaction was stopped by dilution with 40 mL
cyclohexane.
The microparticles
thus formed were separated by centrifugation (5 min, 3000 rpm), washed
successively with cyclohexane, a 95% ethanol solution containing 2% (v/v)
polysorbate 20, 95% ethanol and finally with distilled water.
In the last step,
the colloidal suspension obtained was freeze-dried (Lyophilisator Virtisâ Advantage, vacuum <200 mTorr,
condenser <-40°C, plate +30°C).
Influence of
different parameters was studied, introducing variations in the standard
procedure : amount of monomer, NaOH concentration, reaction time, agitation
rate, amount of cross-linking agent and amount of surfactant. All formulations
were prepared three times.
Physicochemical
characterization of the particles
Influence of these parameters
was studied controlling the size of the particles formed by a laser diffraction
technique (granulometer Coulter LS 100,
IR analysis
The FT-IR spectra acquired
were taken from dried samples. A FT-IR (Spectrum One®, Lita
detector, MIR source and FT-IR Spectrum® software from Perkin Elmer)
was used for the analysis in the frequency range between 4000 and 600 cm-1,
a 8 cm-1 resolution and a 0.2 cm-1 rate. The results were
the means of 16 determinations. Physical mixtures of microparticles and active
drug (1/1) were used as blanks.
Differential scanning
calorimetry (DSC)
Thermal analysis was performed
using a DSC 6 calorimeter equipped with a Pyris® software (Perkin
Elmer). All samples were heated at a 10°C min-1 scanning rate
between 30-350°C after a 1 min stabilization plate at 30°C/20 mW, under
nitrogen atmosphere. Thermograms are expressed in °C = f(mW). Physical mixtures
of b-CD microparticles
with amikacin (1/1) are used as blanks.
Encapsulation
of hydrosoluble substances
Amikacin sulfate was
encapsulated at two different steps of the fabrication :
-encapsulation during the
microparticles fabrication process (solubilization, emulsification,
cross-linking, freeze-drying),
- Encapsulation with the synthesized microparticles
controlling the contact time and the influence of amikacin sulfate amount.
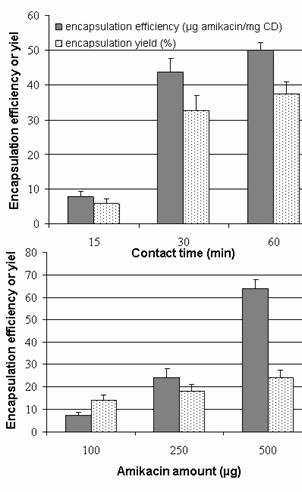
Influence of contact time
A series of experiments were
conducted increasing contact time (15-60 min) between colloidal suspension (100
mL) and amikacin
amount (500 mg). Results show
that equilibrium is reached rapidly, the plate being reached after 30 min
incubation time (Figure11).
Influence of amikacin sulfate
The volume of colloidal
suspension is maintained at 50 mL, with a contact
time fixed at 60 min. Encapsulation efficiency and yield increase linearly with
the amount of amikacin introduced (Figure 11).
Statistical
Analysis
Statistical data analyses were
performed using the Student’s t-test at p < 0.05.
Results and discussion
Microparticles
synthesis
Influence of reactions
parameters on the size of microparticles
Elaboration of 5 mm microparticles
size by interfacial polycondensation of b-CD with the cross-linking
agent, terephtaloyle chloride, was undertaken and assayed varying different
parameters, from the standard procedure.
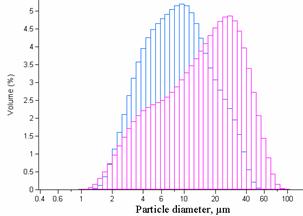
Figure 2. Granulometric features of the obtained
particles by the standard method with 0.5 M (right) and 1 M NaOH (left) (b-CD: 7.5%w/v, 1M NaOH, 30 min reaction time,
8000 rpm agitation rate, polymerization and surfactant agent: 5% w/v). For
unimodal repartition (1M NaOH: n=3, mean=8.549±2.089mm).
The size of the
particles formed increased respectively with the b-CD amount, with NaOH
concentration (Table 1), only with concentration above 1M and the step of
freeze-drying. NaOH concentration lower than 1 M produced a bimodal repartition
of the particles size (Figure 2). Freeze-drying increased the median
microspheres size from 8.7 to 15.3 mm, in a bimodal
repartition (Figure 3) modifying the studied population granulometry features
due to particles agglomeration. On contrary, the size of the particles was
minimal at 5-10% (v/v) of surfactant and at maximal agitation rate (24000 rpm)
(Table 1). The surfactant amount and the agitation rate were determinant in the
emulsification step and crucial for the formation of the smallest particles. An
optimized method was elaborated, modifying the agitation speed to 24 000 rpm
and decreasing the concentration of cross-linking agent to 4.5% (w/v), a concentration
that don't affect the microspheres size, yield and unimodal repartition. This
new preparation method allowed the formation of particles with size inferior to
5 mm (Figure 4) with a
high reproductibility.
Stability assay, on
the form of colloidal suspension were undertaken at ambiant temperature and at
4°C. Results obtained showed a physical stability of uncharged particles for at
least three months at ambiant temperature. At lower temperatures, conservation
of colloidal suspension was more difficult due to an increased sensitivity to
crystallization phenomenon.
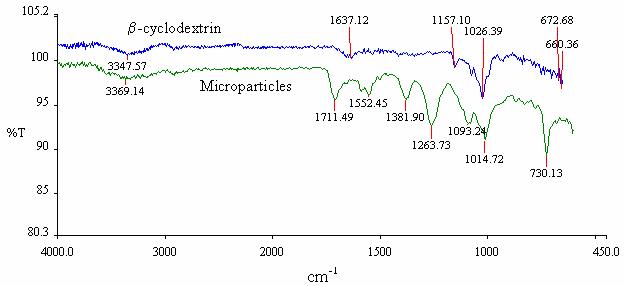
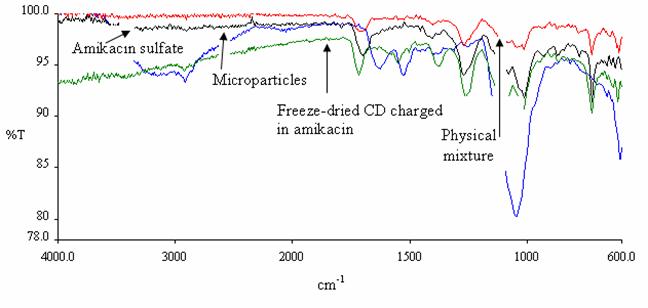
FT-IR spectra
The spectrum of microparticles
formed with the optimized method was compared with the spectrum of original b-CD. Three bands
appeared in microparticle spectrum at 1711, 1263 and 730 cm-1, due
to the formation of esters from hydroxy groups of the b-CD (Figure 5). The mechanisms
have been studied by reaction of b-CD treated with 1
equivalent of terephtaloyle chloride in pyridine (37).
Table 1. Influence of preparation parameters
on mean microparticle size, colloidal suspension pH and yield, when varied
independently from the standard formulation (b-CD: 7.5%w/v, 1M NaOH, 30 min
reaction time, 8000 rpm agitation rate, polymerization and surfactant agent: 5%
w/v) (n=3). The optimized formulation is shown in bold.
|
Parameter |
Value |
pH |
Mean size (mm) ±SD |
Mean yield (%) ±SD |
|
Monomer amount (% w/v) |
5 |
2.68 |
7.89±1.10 |
26.7 ± 3.3 |
|
7.5 |
2.74 |
6.31±1.23 |
26.5± 3.0 |
|
|
10 |
2.89 |
8.92±1.20 |
25.5 ± 3.1 |
|
|
20 |
2.67 |
10.64±0.51 |
27.7 ± 2.9 |
|
|
30 |
2.95 |
12.78±0.45 |
28.4 ± 2.8 |
|
|
NaOH (M) |
0.5 |
1.95 |
15.78±0.97 |
18.1 ± 2.5 |
|
1 |
2.69 |
8.52±1.12 |
28.9 ± 3.5 |
|
|
2 |
3.80 |
10.82±0.74 |
34.7 ± 2.6 |
|
|
4 |
5.60 |
12.79±0.88 |
53.1 ± 4.2 |
|
|
Reaction time (min) |
15 |
2.84 |
17.19±1.51 |
27.5 ± 2.5 |
|
30 |
2.70 |
8.43±0.57 |
29.4 ± 2.1 |
|
|
60 |
2.64 |
8.38±0.35 |
32.8 ± 2.4 |
|
|
120 |
2.95 |
8.71±0.52 |
35.5 ± 2.6 |
|
|
240 |
2.67 |
8.11±0.76 |
36.6 ± 2.5 |
|
|
Surfactant amount (% v/v) |
2.5 |
2.68 |
12.97±0.81 |
24.9 ± 2.4 |
|
5 |
2.71 |
8.25±0.42 |
26.8 ± 2.9 |
|
|
10 |
2.61 |
8.59±0.59 |
27.9 ± 2.5 |
|
|
Cross-linking agent (% w/v) |
1.25 |
6.73 |
13.48±0.61 |
18.8 ± 1.5 |
|
2.5 |
5.10 |
9.56±0.52 |
21.9 ± 2.8 |
|
|
3.75 |
3.90 |
8.56±0.53 |
29.9 ± 2.7 |
|
|
5 |
2.69 |
8.69±0.46 |
28.7 ± 3.4 |
|
|
6.25 |
1.91 |
8.98±0.47 |
27.4 ± 2.6 |
|
|
7.5 |
1.56 |
8.68±0.41 |
28.6 ± 2.5 |
|
|
8.75 |
1.69 |
8.90±0.34 |
29.5 ± 2.4 |
|
|
10 |
1.09 |
8.80±0.46 |
27.5 ± 2.7 |
|
|
Agitation rate (rpm) |
8,000 |
2.64 |
8.68±0.65 |
25.6 ± 2.7 |
|
9,500 |
2.79 |
8.29±0.74 |
27.9 ± 2.5 |
|
|
13,500 |
2.84 |
6.15±0.34 |
28.1 ± 3.5 |
|
|
24,000 |
2.83 |
4.54±0.22 |
29.8 ± 1.2 |
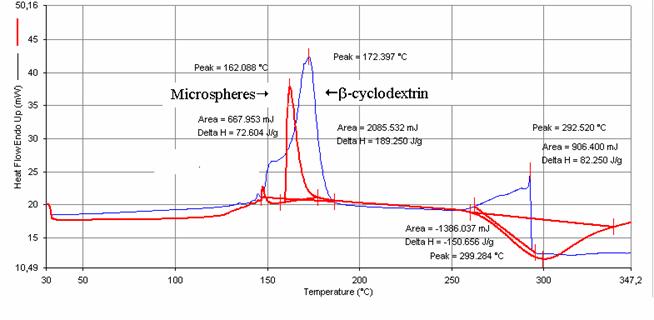
DSC
No observable signal was
present in the temperature range 30-115°C (Figure 6). At 172.4°C, an endotherm
peak responsible for the cyclodextrin melting was observed whereas it was
displaced at 162°C for reticulated cyclodextrin. At 300°C, an additional
exothermic peak was attributed to terephtaloyle chloride.
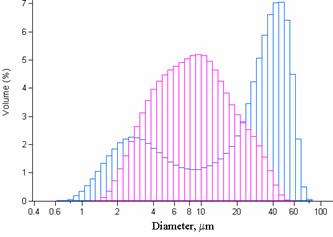
Figure 3. Granulometric features of the obtained
particles by the standard method before freeze-drying step (unimodal) and after
freeze-drying step (bimodal) (b-CD: 7.5%w/v, 1M NaOH, 30 min
reaction time, 8000 rpm agitation rate, polymerization and surfactant agent: 5%
w/v). For unimodal repartition (before freeze-drying: n=3, mean=8.549±2.089mm).
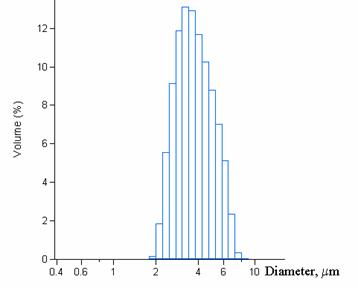
Figure 4. Granulometric features of the obtained
particles for the optimized formulation (n=5 ; pH = 3.65±0.13; size=3.72±1.34 mm ; yield =28.5±1.4%) (b-CD: 7.5%w/v, 1M NaOH, 30 min
reaction time, 24000 rpm agitation rate, polymerization agent: 4.5% w/v and
surfactant agent: 5% w/v).
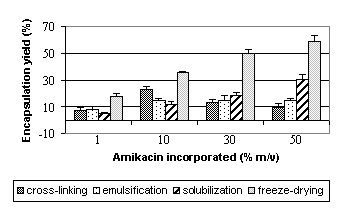
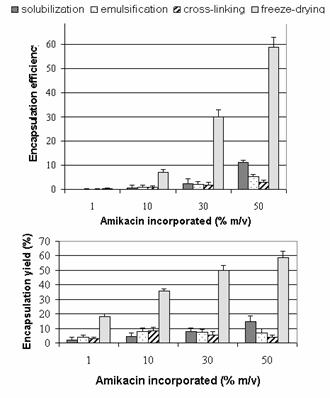
Amikacin sulfate encapsulation
in b-CD microparticles
Encapsulation during
microparticles fabrication process
Encapsulation efficiency (mass
of amikacin sulfate in particles (mg)/ mass of b-CD (mg)) (Figure 7)
increased linearly with the amikacin sulfate incorporated due to the fact that b-CD must be in
excess over the amikacin sulfate incorporated. Encapsulation was more
pronounced for solubilization and freeze-drying step. Encapsulation yield (mass
of amikacin sulfate in particles (mg)*100/mass of
amikacin sulfate introduced (mg)) (Figure 7)
increased for the step of solubilization and freeze-drying reaching a plate
from 20 mg amikacin. On
contrary, yields were constant from 10 mg for emulsification step and
even decreased from 10 mg for cross-linking
step.
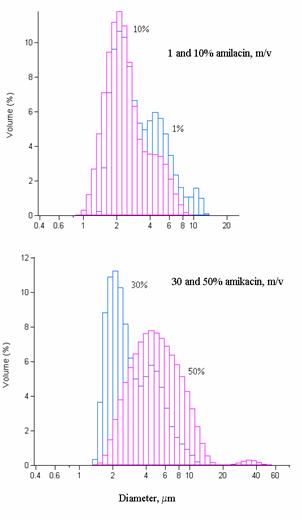
Encapsulation with
microparticles
Ganulometry measurements were
represented in function of% (m/v) amikacin introduced in the microsphere
solution (Figure 8). Until 50% (m/v) amikacin, a bimodal repartition of the
complexed amikacin was observed with a maximum size of particles around 3 mm (1% 3.136±1.689 mm ; 10% 2.433±1.541
mm ; 30% 2.970±1.603
mm). When 50%
amikacin was put into contact with microspheres, a maximum size centered on
4.968±1.689 mm with an unimodal
repartition was obtained reaching a limit for pulmonary drug delivery at
alveolar scale.
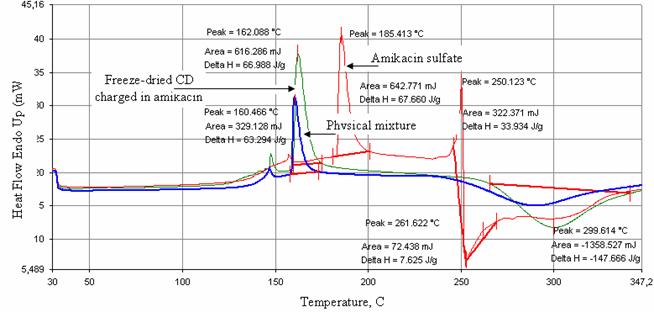
Freeze-drying of a b-CD colloidal
suspension and amikacin sulfate gave a chemical entity whose thermic features
superpose to the host particle (Figure 9).
The thermogram
(Figure 9) and IR spectra (Figure 10) of amikacin charged freeze-dried
cyclodextrin and that corresponding to physical mixture compared with amikacin
sulfate did not show the reapparition of the signal characteristic of amikacin
sulfate. Therefore, this phenomenon can be attributed to dilution of the active
substance in the powder. The nature of the interactions between amikacin and
cyclodextrins remains unknown. Two hypotheses can be emitted : either the
amikacin molecule forms partially or wholly an inclusion complex with
cyclodextrins or one of the reaction functions of amikacin reacts with hydroxyl
groups from cyclodextrin to create an hydrogen bond.
The first
hypothesis seems unlikely because inclusion complexes are observed with
hydrophobic compounds which insert inside the hydrophobic