J Pharm Pharmaceut Sci
(www.cspscanada.org) 8(3):419-425, 2005
Synthesis and analgesic activity of
N-arylhydrazone derivatives of mefenamic acid
Ali Almasirad, Mohammad Tajik, Davood
Bakhtiari, Abbas Shafiee
Mohammad Abdollahi, Mohammad Jafar zamani,
Reza khorasani, Hadi Esmaily
Department of Medicinal Chemistry, Faculty of Pharmacy, and
Pharmaceutical Sciences Research Center, Tehran University of Medical Sciences,
Tehran, Iran
Department of Toxicology and Pharmacology, Faculty of Pharmacy and Pharmaceutical
Sciences Research Center, Tehran
University of Medical Sciences, Tehran, Iran
Received December
19, 2004; Revised July 23, 2005; Accepted August 15, 2005, Published
September 1,
2005
Corresponding author:
Abbas Shafiee, Department of Medicinal Chemistry, Faculty of Pharmacy, and Pharmaceutical Sciences
Research Center,
Tehran University of Medical Sciences, Tehran,
Iran. ashafiee@ams.ac.ir
PDF Version
Abstract: PURPOSE:
A series of N-Arylhydrazone derivatives of mefenamic acid (a known non-steroidal
anti-inflammatory drug) were synthesized in order to obtain new compounds with potential
analgesic and anti-inflammatory activity. METHODS: The structures of all
synthesized compounds were confirmed by means of infrared, proton magnetic
resonance and mass spectroscopy. All compounds were evaluated for their
analgesic and anti-inflammatory activities by abdominal constriction test
(writhing test) and carrageenan-induced rat paw edema test respectively. RESULTS:
Most of the synthesized compounds induced significant reduction in the writhing
response when compared to control. Among them, compounds 11, 12, 15, 16, 19,
20, and 21 were significantly more potent than mefenamic acid in the
writhing test. The anti-inflammatory activity of these 7 compounds were evaluated and compounds 11, 12, 16,
19 and 20 showed significant anti-inflammatory activity in comparison
to control but their effect was weaker
than mefenamic acid. CONCLUSIONS: The antinociceptive relative activity
of some of these newly synthesized compounds is greater than mefenamic acid but
they are not potent anti-inflammatory agents.
INTRODUCTION
Non-steroidal
anti-inflammatory drugs (NSAIDS) are widely used in the treatment of pain and
inflammation. These compounds non selectively inhibit the two isoforms of the
cyclooxygenase (COX-1 and COX-2) and thus prevent the metabolism of cellular
arachidonic acid (AA) and the upregulation of prostaglandin formation, which
otherwise lead to an increase of vascular permeability, edema, hyperalgesia,
pyrexia and inflammation. In addition to COX, the 5-lipoxygenase (5-LO) enzyme
is another key enzyme which is involved in the AA cascade. Leukotrienes,
produced through the 5-LO enzyme pathway, may also contribute to both
inflammation and NSAIDs induced side effects. For these reasons, compounds that
are dual inhibitors of both COX and 5-LO are being studied as potential
analgesic and anti-inflammatory agents with an improved safety profile in
comparison to NSAIDS (1-2). Currently, various chemical families of dual
COX/5-LO inhibitors can be found in the scientific literatures (3). In the 1980’s,
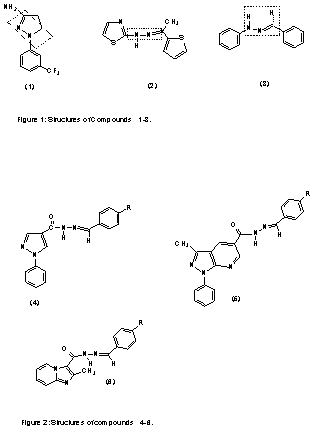
hydrazone-type containing compounds such as BW 755c (1) and CBS 1108 (2)
(Fig 1) were described as dual COX/5-LO inhibitors which present analgesic and
anti-inflammatory activities.
In fact, some evidences suggest that the hydrazone
moiety present in derivative 3 (Fig 1) possess a pharmacophoric
character for the inhibition of COX. According to these results, analgesic
profile of new series of heterocyclic N-acylarylhydrazones 4-6 (Fig 2)
has been previously described (4-5).
In addition to these compounds, there are some reports
about importance of fenamate structures in dual inhibition of COX/5-LO by
substitution of their carboxylic acid moiety with some acidic heterocycles,
namely 1,3,4-oxadiazole-2-thione (7)
and 1,3,4-thiadiazole-2-thione (8) (Fig
3) eg in mefenamic acid (1,6).
Thus, we decided to replace the carboxylic acid moiety
of mefenamic acid, a known NSAID drug, with an N-arylhyrazone group in the hope
of obtaining additional inhibitors of cellular AA metabolism.
MATERIAL AND METHODS
Chemistry
Chemicals
were purchased from Merck Chemical Company (Darmstadt, Germany).
Melting points were taken on a Kofler hot stage apparatus (Reichert, Vienna, Austria)
and are uncorrected. 1H-NMR spectra were obtained using a Brucker
FT-80 spectrometer (Brucker, Rheinstetten,
Germany).
Tetramethylsilane was used as an internal standard. Mass spectra were obtained
using a Finnigan Mat TSQ-70 spectrometer at 70 ev (Finnigan Mat, Bremen, Germany). The IR spectra were
obtained using a Nicolet FT-IR Magna 550 spectrographs (KBr disks) (Nicolet,
Madision, WI, USA).
The purity of compounds was confirmed by TLC using different mobile phases. The
results of the elemental analyses (C, H, N) were within±0.4% of theoretical
values for C, H and N.
Synthesis of N-arylhdyrazone
derivatives of mefenamic acid (11-22).
The
hydrazide derivative of mefenamic acid (9) (Scheme 1) was prepared according
to the previously described methods (7-8).
The
target compounds were synthesized by acid-catalyzed condensation of 9
with the corresponding aromatic aldehydes (Scheme 1) (4).
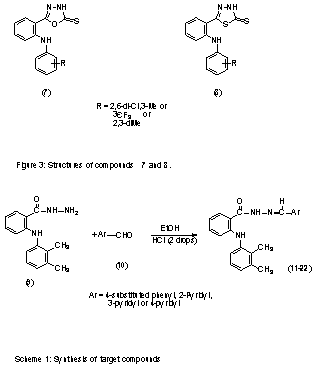
A mixture of 1.9 mmol of hydrazide 9 and 1.9
mmol of the corresponding aldehyde derivative 10 in 20 ml of absolute
ethanol was stirred at room temperature for 0.5 to 1h, in the presence of two
drops of hydrochloric acid as a catalyst. The end of the reaction was observed
by TLC, and the hydrazones 11-22 were isolated by concentration of the
reaction mixture under reduced pressure, followed by neutralization with a 10%
aqueous solution of sodium bicarbonate. The resulting precipitate was filtered,
washed with 10 ml water and crystallized from a suitable solvent. Melting
points, crystallization solvents and yields are reported in
Table 1.
The spectral data of new synthesized compounds are as
follows: Compound 11:- IR (KBr): n cm-1 3319 (NH), 3196 (NH), 1634
(C=0). -1H-NMR (CDCl3): d (ppm) 9.12 (bs, 1H, NH), 9.07 (bs, 1H,
NH), 8.11 (s, 1H, CH), 7.60 (t, 3H, aromatic), 7.14-6.46 (m, 8H, aromatic),
2.37 (s, 3H, CH3), 2.31 (s, 3H, CH3), 2.19 (s, 3H, CH3).-
MS: m/z (%) 357 (M+, 68), 224 (100), 208 (30), 180 (17). Compound
12:- IR (KBr): n cm-1 3345 (NH), 3237 (NH), 1634 (C=0). -1H-NMR
(CDCl3): d (ppm) 9.18 (bs, 1H, NH), 9.08 (bs, 1H, NH), 8.16(s,
1H, CH), 7.82-7.45 (m, 3H, aromatic), 7.19-6.58 (m, 8H, aromatic), 2.32 (s, 3H,
CH3), 2.19 (s, 3H, CH3).- MS: m/z (%) 361 (M+,
67), 224 (100), 207 (21), 179 (19). Compound 13: -IR (KBr) n cm-1
3298 (NH), 3211 (NH), 1629 (C=0). -1H-NMR (CDCl3): d (ppm) 9.13
(bs, 1H, NH), 9.04 (bs, 1H, NH), 8.10 (s, 1H, CH), 7.77-7.50 (m, 3H, aromatic),
7.14-6.73 (m, 8H, aromatic), 3.84 (s, 1H, OCH3), 2.32 (s, 1H, CH3),
2.20 (s, 1H, CH3).-MS: m/z (%), 373 (M+, 48), 222 (100),
207 (20), 180 (11).
Table 1: Physical data of
synthesized compounds
|
Compound
No
|
Ar
|
MP°C
|
yield
%
|
Recrystallization
Solvent
|
Molecular
Formula
|
|
11
|
4-tolyl
|
197-199
|
14
|
EtOAc/Petr-Ether
|
C23H23N3O
|
|
12
|
4-fluorophenyl
|
195-197
|
35
|
EtOH
|
C22H20FN3O
|
|
13
|
4-methoxyphenyl
|
216-217
|
68
|
EtOAc
|
C23H23N3O2
|
|
14
|
phenyl
|
192-193
|
24
|
EtOAc/Petr-Ether
|
C22H21N3O
|
|
15
|
4-hydroxyphenyl
|
190-194
|
27
|
EtOAc
|
C22H21N3O2
|
|
16
|
4-nitrophenyl
|
221-222
|
62
|
EtOH
|
C22H20N4O4
|
|
17
|
4-chlorophenyl
|
204-206
|
43
|
EtOH
|
C22H20ClN3O
|
|
18
|
4-N,N-dimethylamino-phenyl
|
247-248
|
44
|
EtOH
|
C24H26N4O
|
|
19
|
4-pyridyl
|
203-204
|
76
|
EtOAc
|
C21H19N4O
|
|
20
|
3-pyridyl
|
182-185
|
39
|
MeOH
|
C21H19N4O
|
|
21
|
2-pyridyl
|
160-161
|
41
|
EtOAc
|
C21H19N4O
|
|
22
|
4-Bromophenyl
|
212-215
|
90
|
MeOH
|
C22H20BrN3O
|
Compound 14:- IR (KBr): n cm-1 3324 (NH), 3206 (NH), 1629
(C=0). -1H-NMR (CDCl3): d (ppm) 9.32 (bs, 1H, NH), 9.15 (bs, 1H,
NH), 8.17 (s, 1H, CH), 7.76-6.72 (m, 12H, aromatic), 2.32 (s, 3H, CH3),
2.19 (s, 3H, CH3).- MS: m/z (%) 343 (M+, 55), 224 (100), 209
(17). Compound 15:- IR (KBr): n cm-1 3355 (OH), 3288 (NH), 3210
(NH), 1620 (C=0). -1H-NMR (DMSO-d6): d (ppm) 11.05
(bs, 1H, OH), 9.5 (bs, 1H, NH), 9.32 (bs, 1H, NH), 8.33 (s, 1H, CH), 7.58-7.42
(m, 3H, aromatic), 7.21-6.68 (m, 3H, aromatic), 2.31 (s, 3H, CH3),
2.15 (s, 3H, CH3).- MS: m/z (%) 359 (M+, 55), 224 (100),
208 (29), 180 (19). Compound 16:- IR (KBr): n cm-1 3355 (NH), 3280 (NH), 1639
(C=0), 1347 (NO2), 1521 (NO2). -1H-NMR (CDCl3):
d (ppm) 9.46
(bs, 1H, NH), 9.16 (bs, 1H, NH), 8.30 (s, 1H, CH), 8.24 (d, J=8.8 Hz, 2H, aromatic),
7.89 (d, J=8.8 Hz, 2H, aromatic), 7.55 (dd, J=7.9, 1.5 Hz, 1H, aromatic), 7.18-6.61
(m, 6H, aromatic), 2.32 (s, 3H, CH3), 2.19 (s, 3H, CH3).-
MS: m/z (%) 388 (M+, 48), 222 (100), 208 (32), 180 (16). Compound
17:- IR (KBr): n cm-1 3329 (NH), 3196 (NH), 1629 (C=0). -1H-NMR
(CDCl3): d (ppm) 9.32 (bs, 1H, NH), 9.03 (bs, 1H, NH), 8.15 (s,
1H, CH), 7.73-6.69 (m, 11H, aromatic), 2.31 (s, 3H, CH3), 2.19 (s,
3H, CH3).- MS: m/z (%) 377 (M+, 19), 222 (100), 207 (25),
180 (14).
Compound 18: -IR (KBr): n cm-1 3240 (NH), 3211 (NH), 1634
(C=0).-1H-NMR (CDCl3): d (ppm) 9.10 (bs, 1H, NH), 8.95 (bs, 1H,
NH), 8.03 (s, 1H, CH), 7.70-7.45 (m, 3H, aromatic) 7.15-6.64 (m, 8H, aromatic),
3.02 (s, 6H, N(CH3)2), 2.31 (s, 3H, CH3), 2.20
(s, 3H, CH3). –MS: m/z (%) 386 (M+, 81), 223 (74), 163
(100), 40 (38). Compound 19: -IR (KBr): n cm-1 3355 (NH), 3217 (NH), 1639
(C=0). -1H-NMR (CDCl3): d (ppm) 9.52 (bs, 1H, NH), 9.04 (bs, 1H, NH),
8.66 (d, 2H, J=6Hz, aromatic), 8.21 (s,1H, CH), 7.64-7.53 (m, 3H, aromatic)
7.15-6.70 (m, 6H, aromatic).- MS: m/z (%) 344 (M+, 28), 224 (100),
209 (27), 180 (22). Compound 20: -IR (KBr): n cm-1 3331(NH), 3192 (NH), 1625
(C=0). -1H-NMR (CDCl3): d (ppm) 9.78 (bs, 1H, NH), 9.05 (bs, 1H,
NH), 8.78 (s, 1H, aromatic), 8.60 (d, J=6.2 Hz, 1H, aromatic), 8.27 (s, 1H, CH)
8.13 (d, J=6.2 Hz, 1H, aromatic), 7.58 (d, J=7.9 Hz, 1H, aromatic), 7.37-6.67
(m, 7H, aromatic), 2.31 (s, 3H, CH3), 2.17 (s, 3H, CH3).-
MS: m/z (%) 344 (M+, 57), 224 (100), 209 (29), 180 (26), 120 (16). Compound
21: -IR (KBr): n cm-1 3283 (NH), 3190 (NH), 1632 (C=0). -1H-NMR
(CDCl3): d (ppm) 9.41 (bs, 1H, NH), 9.20 (bs, 1H, NH), 8.71-8.47 (m, 3H, aromatic), 8.22 (s,
1H, CH), 7.95-6.55 (m, 8H, aromatic), 2.28 (s, 3H, CH3), 2.14 (s,
3H, CH3). –MS: m/z (%) 344 (M+, 95), 224 (100), 208 (22),
180 (19), 120 (68), 92 (40).
Compound 22: -IR (KBr): n cm-1 3331 (NH), 3169 (NH), 1622
(C=0).-1H-NMR (CDCl3): d (ppm) 11.55 (bs, 1H, NH), 9.21 (bs, 1H,
NH), 8.37 (s, 1H, CH), 7.72-7.43 (m, 5H, aromatic) 7.28-6.56 (m, 6H, aromatic),
2.31 (s, 3H, CH3), 2.19 (s, 3H, CH3). –MS: m/z (%) 422 (M+,
21), 223 (100), 210 (28), 180 (25).
Pharmacology
Male
NMRI mice weighing 20-25 g and male Wistar rats weighing 200-250 g (from animal
house of Faculty of Pharmacy, TUMS) were used for the abdominal constriction
test (writhing test) and the carrageenan-induced paw edema respectively. The
animals were housed in colony cages and conditions of constant temperature (22
± 2°C) and a 12 h light/dark schedule and allowed free
access to standard diet and tap water except during the experiment. The animals
were allowed to habituate to the laboratory environment for 2h before the
experiments were initiated. All ethical manners for use of laboratory animals were
considered carefully and the protocol of study was approved by TUMS ethical
committee. The compounds were administered intraperitoneally (IP) (31 mmol/kg; 0.2
ml/20g) as a suspension in saline and tween 80 (4% w/n). Mefenamic acid (Hakim Pharmaceutical Co)
(31 mmol/kg, IP) (9) was used as standard drug under the
same conditions. The control group received vehicle (0.2 ml/20g, IP) alone.
Analgesic Activity
The
analgesic activity was determined in vivo by the abdominal constriction test
induced by acetic acid (0.6%; 0.1 ml/10g) in mice (10). An acetic acid solution
was administered IP 30 minutes after administration of compounds.
Antinociception was recorded by counting the number of writhings immediately
after injection of acetic acid during 30 minutes. The analgesic activity was
expressed as the percentage of inhibition of constrictions when compared with
the vehicle control group Table 2.
Anti-inflammatory activity
The
anti-inflammatory activity was determined in vivo using the carrageenan-induced
rat paw edema test (5, 11). A solution of 0.1 ml of 1% carrageenan
(Sigma-Aldrich, Dorset, UK) in saline was injected sub plantarly in the right
hind paw of the rats 1h after IP administration of compounds. The paw thickness
was measured from the ventral to the dorsal surfaces using a dial caliper
immediately prior to carrageenan injection and then at each hour, up to 4 h
after the sub planar injection. The edema was calculated as the thickness
variation between the carrageenan and saline treated paw. Anti-inflammatory
activity was expressed as the percent of inhibition of the edema when compared
with the control group.
Statistics
The results are expressed as the mean ± SEM of n
animals per group. The data were statistically analyzed by one way analysis of Variance
(ANOVA) followed by Tukey multicomparison test. differences with P<0.05
between experimental groups were considered statistically significant.
RESULTS
All
new N-arlyhydrazone derivatives of mefenamic acid (11-22) were initially
evaluated for analgesic activity using the acetic acid induced mice abdominal
constrictions test and the results are shown in
Table 2.
Except compounds 14,
17, 18 and 22, all of them
induced significant reduction in the writhing response in comparison to
control and among them 7 compounds
significantly showed higher inhibitory effect on the writhing response in
comparison to mefenamic acid as follow: (11,
71.1%, P<0.001) (12, 58.5%,
P<0.001), (15, 46.7%, P<0.01),
(16, 42.4%; P<0.01), (19, 67.9%; P<0.001), (20, 93.7%, P<0.001), (21,
50.9%, P<0.01), mefenamic acid (25.6%, P<0.01).
Table 2: Effects of Compounds 11-22 and
mefenamic acid in the abdominal constrictions induced by acetic acid in mice
|
Compound
|
Dose
(mmol/kg)1
|
Constriction
No.
(mean
± SEM)
|
Inhibition
(%)2
|
Relative
activity3
|
P
value
|
|
Control
|
-
|
70.33
± 3.00
|
-
|
-
|
-
|
|
mefenamic
acid
|
31
|
52.33
± 2.59
|
25.59
|
1
|
P<0.01
vs. control
|
|
11
|
31
|
20.33
± 4.62
|
71.09
|
2.78
|
P<0.001 vs. control
P<0.001 vs. ma4
|
|
12
|
31
|
29.20
± 3.73
|
58.45
|
2.28
|
P<0.001 vs. control
P<0.001 vs. ma
|
|
13
|
31
|
47.50
± 4.37
|
32.45
|
1.27
|
P<0.01 vs. control
P>0.05 vs. ma
|
|
14
|
31
|
65.00
± 4.54
|
7.57
|
0.30
|
P>0.05 vs. control
P<0.05 vs. ma
|
|
15
|
31
|
37.50±2.32
|
46.67
|
1.82
|
P<0.001 vs. control
P<0.01 vs. ma
|
|
16
|
31
|
40.50±2.59
|
42.41
|
1.66
|
P<0.001 vs. control
P<0.01 vs. ma
|
|
17
|
31
|
65.17±5.11
|
7.34
|
0.29
|
P>0.05 vs. control
P<0.05 vs. ma
|
|
18
|
31
|
66.00±3.28
|
6.16
|
0.24
|
P>0.05 vs. control
P<0.05 vs. ma
|
|
19
|
31
|
22.60±4.17
|
67.86
|
2.24
|
P<0.001 vs. control
P<0.001 vs. ma
|
|
20
|
31
|
4.4±2.01
|
93.70
|
3.66
|
P<0.001 vs. control
P<0.001 vs. ma
|
|
21
|
31
|
34.50±5.57
|
50.94
|
1.99
|
P<0.001 vs. control
P<0.01 vs. ma
|
|
22
|
31
|
71.33±5.57
|
-1.42
|
-5.5
|
P>0.05 vs. control
P<0.05 vs. ma
|
1
number of animals in each group n= 6;2 % inhibition obtained by comparison with
vehicle control group; 3 Analgesic activity relative to mefenamic
acid 4 ma (mefenamic acid).
Since these 7 compounds showed more analgesic activity
in comparison to mefenamic acid, we decided to evaluate their anti-inflammatory
profile by using the carrageenan induced rat paw edema test and the results are
summarized in
Table 3.
Compounds 11,
12, 16 and 19 as well as
mefenamic acid as the reference drug induced significant anti-inflammatory
activity after 3 and 4 h in comparison to control but none of the tested
compounds was more active than mefenamic acid. Compound 20 showed significant anti-inflammatory activity after 1, 2 and 3h
in comparison to control and after 1 h in comparison to mefenamic acid.
DISCUSSION
The
pharmacological results of the present study show a good analgesic profile in
comparison to control and mefenamic acid. Similar to the previous studies the
most active derivatives 19, 20 and 21 possess the pyridine
ring at the aryl moiety of the arylhydrazone frame work (4). In addition the
compounds possessing the 4-tolyl or 4-flurophenyl moiety 11, 12
respectively are among the most active compounds in our study. Previous studies
indicated that 4-Bromophenyl and 4-N, N-dimethyl-aminophenyl have a major
contribution to the analgesic activity (4-5,
12-13) but our pharmacological evaluations showed opposite results.
Compounds 18, 22 are among the weakest synthesized structures. As we described
some N-arylhydrazone derivatives 4-6
(Fig 2) have presented an important analgesic profile which found to be more
influenced by the nature of phenyl ring substituent of the hydrazone sub-unit
than the pattern of the heterocyclic ring of the N-acyl moiety (4). Therefore,
it is possible that replacement of these kinds of acyl groups with a fenamate
structure has changed the mechanism of enzyme-receptor interaction and the importance
of 4-substituents of phenyl rings at the aryl moiety of the aryl hydrazone
frame work. Since in vivo activity depends on highly complex physiological
interactions, therefore at this moment we are unable to rationalize all of
these pharmacological results.
Table 3: Effects of compounds (11, 12, 15, 16, 19, 20, 21)
and mefenamic acid in the inhibition of carrageenan-induced rat paw edema
|
Compound
|
Time
(h)1
|
Dose
(mml/kg)2
|
Thickness variation
(mm)3
|
Inhibition
(%)4
|
p value5
|
|
Control
|
1
2
3
4
|
-
-
-
-
|
1.062±0.123
2.072±0.157
2.748±0.161
2.413±0.300
|
-
-
-
-
|
-
-
-
-
|
|
mefenamic acid
|
1
2
3
4
|
31
31
31
31
|
1.00±0.118
1.937±0.258
1.412±0.300
0.915±0.202
|
5.8
6.5
48.6
62
|
P>0.05
P>0.05
P<0.01
P<0.01
|
|
11
|
1
2
3
4
|
31
31
31
31
|
1.497±0.289
1.817±0.271
1.697±0.297
1.370±0.351
|
-40.9
12.3
38.2
43.2
|
P>0.05
P>0.05
P<0.05
P<0.05
|
|
12
|
1
2
3
4
|
31
31
31
31
|
1.327±0.098
2.173±0.183
1.865±0.069
1.265±0.250
|
-24.9
-4.9
32.1
47.57
|
P>0.05
P>0.05
P<0.001
P<0.05
|
|
15
|
1
2
3
4
|
31
31
31
31
|
0.910±0.187
1.770±0.137
2.265±0.172
2.212±0.329
|
14.3
14.57
17.57
8.33
|
P>0.05
P>0.05
P>0.05
P>0.05
|
|
16
|
1
2
3
4
|
31
31
31
31
|
1.525±0.229
2.635±0.244
1.665±0.178
1.450±0.201
|
-43.6
-27.2
39.4
39.9
|
P>0.05
P>0.05
P<0.01
P<0.05
|
|
19
|
1
2
3
4
|
31
31
31
31
|
1.120±0.218
1.770±0.069
1.723±0.157
1.480±0.129
|
-5.5
14.3
37.3
38.67
|
P>0.05
P>0.05
P<0.01
P<0.05
|
|
20
|
1
2
3
4
|
31
31
31
31
|
0.512±0.105
1.330±0.237
1.960±0.213
2.250±0.336
|
51.8
35.8
28.67
6.75
|
P<0.01
P<0.05
P<0.05
P>0.05
|
|
21
|
1
2
3
4
|
31
31
31
31
|
1.375±0.131
2.433±0.183
2.495±0.131
2.067±0.148
|
-29.47
-17.42
9.2
14.3
|
P>0.05
P>0.05
P>0.05
P>0.05
|
1
Time after carrageenan injection (0.1 mg/paw); 2 number of animals
in each group n=6; 3 Thickness variation is the difference between
the thickness of carrageenan-treated paw and saline-treated paw; 4
percentage of inhibition obtained by comparison with the vehicle; 5
p value of all compounds except the first hour of compound 20, the third and
fourth hours of compound 21 in comparison to mefenamic acid was
P>0.05; results are expressed as mean ± SEM.
The anti-inflammatory evaluation of 7 most potent
compounds showed that replacement of carboxylic acid group of mefenamic acid
with N-arylhydrazone moiety can not produce any advantage in the
anti-inflammatory properties of this drug. Most of the synthesized compounds
had a similar bioavailability profile to mefenamic acid because this reference
drug and compounds 11, 12, 16 and 19 were active
after 3 and 4 h in comparison to control. Compound 20 showed significant
anti-inflammatory effect only after 1h in comparison to mefenamic acid and in
the first 3 h in comparison to control. The effect of compound 20
decreased stepwise from the first hour to the forth hour. Therefore in spite of
a relative high potency after 1h it does not have a good kinetic profile.
We can
deduce from the results that replacement of the acidic moiety of mefenamic acid
with N-arylhydrazone moiety can create potent analgesic compounds. Further
studies are needed to explore the differences in the efficacy and safety of
synthesized compounds.
Acknowledgement
This work was supported by grants from the research
council of Tehran University of Medical Sciences and Iran Chapter of TWAS.
REFERENCES
[1] Charlier, C. and Michaux, C., Dual
inhibiton of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) as a new
strategy to provide safer non-steroidal anti inflammatory drugs. Eur J Med
Chem, 38: 645-659, 2003.
[2]
Unangst, P. C. Connor, D. T. Centenko, W. A. Sorenson, R. J. Kostlan, R. K.
Sircar, J. C. Wright, C. D. Schrier, D. J. and Dyer, R. D., Synthesis and
biological evaluation of 5-[[3, 5-Bis (1, 1-dimethylethyl)-4-hydroxyphenyl]
methylene] oxazoles,-thiazoles and imidazoles: Novel dual 5-lipoxygenase and
cyclooxygenase inhibitors with anti inflammatory activity. J Med Chem,
37 322-328, 1994.
[3]
de Leval, X. Delarge, J. J. Pirotte, B. and Dogne, J. M., New trends in dual 5
LOX/COX inhibition. Curr Med Chem, 9: 941-962, 2003.
[4] Lima, P. C. Lima, L.M. da Silva, K. C. M.
Leda, P. H. O. de Miranda, A. L. P. Fraga, C. A. M. and Barreiro, E. J., Synthesis and analgesic
activity of novel N- acylarylhydrazones and isosters, derived from natural
safrole. Eur J Med Chem,
35: 187-203, 2000.
[5]
Figueiredo, J. M. Camara, C. A. Amarante, E. G. Miranda, A. L. P. Santos, F. M.
Radrigues, C. R. Fraga, C. A. M. and Barreiro, E. J., Design and synthesis of
novel potent antinociceptive Agents: Methyl-imidazolyl N-Acylharazone
derivatives. Bioorg Med Chem, 8: 2243-2248, 2000.
[6]
Boschelli, D. H. Connor, D. T. Bornemeier, D. A. Dyer, R. D. Kennedy, J. A.
Kuipers, P. J. Okonkwo, G. C. Schrier, D. J. and Wright, C. D.,
1,3,4-oxadiazole, 1, 3, 4-thiadiazole, and 1, 2, 4-triazole analogs of the
fenamates: In vitro inhibition of cyclooxygenase and 5-lipoxygenase Activities.
J Med Chem, 36: 1802-1810, 1993.
[7]
Belliotti, T. R. Xostlan, C. R. Kramer, J. B. and Sircar, J. C., Preparation of
N- (fluorenylmethyl)-N-hydroxy and alkanoides and analogs as inflammation
inhibitors. US. Patent 4,981,865.
1991; Chem Abstr 1991, 114, P 206798x.
[8]
Alfred, S. Fitzi, K. and Pfister, R., Analgesic cinnamic acid derivatives. South
African Patent 6, 705, 990. 1963; Chem Abstr, 1969, 70, p 67938a.
[9]
Villa Senor, I. M. Gajo, R. M. T. and Gonda, R. C., Bioactivity studies on the
alkaloid extracts from seeds of leucaena leucocephala. Phytother Res,
11: 615 617, 1997.
[10]
Whittle, B. A., The use of changes in capillary permeability in mice to distinguish between narcotic and non narcotic
analgesics. Br J Pharmacol, 22: 246-253, 1964.
[11]
Al-Haboubi, H. A. Zeitlin, I. J., Re-appraisal of the role of histamine in
carrageenan induced oedema. Eur J Pharmacol, 88: 160-176, 1983.
[12]
Ribeiro, I. G. da Silva, K. C. M. parrini, S. C. de Miranda, A. L. P. Fraga, C.
A. M. Barreiro, E. J., Synthesis and antinociceptive properties of new
structurally planned imidazo [1, 2-a] pyridine 3-acylarylhydrazone derivatives.
Eur J Med Chem, 33: 223-235, 1998.
[13] Leite, L. F. C. C. Ramos, M. N. da Silva, J. B.
P. Miranda, A. L. P. Fraga, C. A. M. and Barreiro, E. J., Synthesis and analgesic profile of novel
N-containing heterocycle derivatives: arylidene 3-phenyl-1, 2,
4-oxadiazole-5-carbohydrazide. IL Farmaco, 54: 747-757, 1999.
JPPS Contents
Published by the Canadian Society
for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian
Society for Pharmaceutical Sciences.
http://www.cspscanada.org/
CSPS Home |
JPPS Home |
Search |
Subscribe to JPPS