J Pharm Pharmaceut Sci (www.cspscanada.org) 8(2):259-271, 2005
Emerging Pharmacological Approaches to the Treatment of Obesity
Kishor M. Wasan* and Norbert A. Looije
Received April 25, 2005, Revised, May 16, 2005, Accepted May 19, 2005, Published, August 5, 2005
ABSTRACT. The obesity epidemic has been recognized by the World Health Organization (WHO) as one of the top 10 global health problems. Worldwide, more than one billion adults are overweight and over 300 million are obese. The majority of developed countries, including the United States, Canada and England are experiencing dramatic increases in obesity. Obesity is a condition associated with the accumulation of excessive body fat resulting from chronic imbalance of energy whereby the intake of energy exceeds expenditure. The excess body fat predisposes an obese individual to chronic diseases, such as coronary heart disease, type 2 diabetes and diseases of the gall bladder and cancer. The high incidence of obesity and the lack of safe pharmaceutical agents have fuelled an increase in anti-obesity drug-related research. Although a number of pharmacological approaches have been investigated in recent years, few safe, therapeutically effective products have been developed. This commentary focuses on emerging pharmacological approaches targeted for the treatment of obesity.
Abbreviations:
ALT, alanine aminotransferase; AMPK, 5’-AMP-activated protein kinase; AST, aspartate aminotransferase; DP-IV, Dipeptidyl peptidase IV; EE, energy expenditure; FDA, Food and Drug Administration US; FM-VP4, Disodium ascorbyl phytostanyl phosphates; GIP, gastric inhibitory compound; GLP, glucagons-like peptide; HDL, high-density lipoprotein; LDL, low-density lipoprotein; NPY, neuropeptide Y; OEA, oleoylethanolamide; PTP1B, protein tyrosine phosphatase 1B; PPAR, peroxisome proliferative activated receptor; PYY, pancreatic peptide Y; SCD-1, stearoyl-CoA desaturase-1; TG, triglycerides; TNF, tissue necrosis factor; TZD, thiazolidinediones; UCP, uncoupled proteins.
What is obesity and why is treatment necessary?
Obesity is defined as a condition associated with the accumulation of excessive body fat resulting from chronic imbalance of energy whereby the intake of energy exceeds expenditure. Excess body fat predisposes an individual to chronic diseases, such as coronary heart disease, type 2 diabetes, diseases of the gall bladder and cancer, and joint degeneration (1,2). The high incidence of obesity, its impact on life expectancy in adults and children, and the lack of safe therapeutic agents, have fuelled an increase in anti-obesity drug-related research. For the first time in years scientists are reporting that life expectancy in adults and children are now reduced due to obesity (6). These latest findings are yet another warning that our current lifestyle of processed foods and decreased physical activity may bring a day when children will have shorter life span than their parents. Although a number of pharmacological approaches have been investigated in recent years, few therapeutically effective and safe products have been developed (3).
Genetic Aspects of Obesity
Thrifty Gene Theory of Obesity
The concept of a "thrifty genotype" was first proposed nearly 50 years ago (4). It was proposed that certain genotypes were selected for survival of the organism. A "thrifty" genotype was defined as "being efficient in the intake and/or utilization of food." Thus, during famines, individuals with a "thrifty" genotype would have a survival advantage because they relied on larger, previously stored energy to maintain homeostasis, whereas those without "thrifty" genotypes would be at a disadvantage and less likely to survive (4). In a society where access to food is no longer an issue and where indeed there is commercial encouragement (i.e. the “Supersized marketing approach) to overeat, such thrifty genes transition from being prosurvival to being detrimental in the development of obesity (4). Chakravarthy and Booth (5) have extended the thrifty gene hypothesis suggesting that survival during the feast-famine cycle the hunter-gatherer selected genes to support a "physical activity cycle" in which cycling of metabolic processes was triggered by the reduction of skeletal muscle glycogen and triglyceride stores. It is further speculated that some "thrifty" gene and genotypes were selected to support required physical activity for survival as well as being implicated in the accumulation and storage of fat.
Role of Leptin, Insulin and Appetite-regulating hormones in the development of obesity
Current research has focused on understanding the molecular signals used to regulate both long- and short-term changes in weight (7-12). Administration of the horomone leptin to obese animals produced weight loss by decreasing appetite while at the same time increasing the rate of fat metabolism. Leptin activates 5’-AMP-activated protein kinase (AMPK) in muscle, inhibiting acetyl coenzyme A, an enzyme that catalyzes a key step in fat synthesis (Figures 1 and 2) (13-16). As a result, the energy sources that go into fat formation are shifted into an oxidative pathway providing energy for muscle cells. In liver, leptin turns down the activity of the gene for stearoyl-CoA desaturase-1 (SCD-1) which has a similar role to acetyl CoA (17-20). Leptin-deficient mice can be protected from obesity by inactivating the SCD-1 gene.
Inhibition of insulin action in mouse brain by knocking out the insulin receptors led to animals overeating and becoming fat (21). Insulin infused into the brain near the arcuate nucleus inhibited production of the appetite-stimulating neuropeptide Y (NPY) (22) (Figure 1). Inhibition of insulin receptor production in the arcuate nucleus of mice resulted in an increase in food intake (22). Knock out of the enzyme protein-tyrosine phosphatase 1B (PTP-1B) that is , responsible for inhibiting leptin and insulin signaling in the hypothalamus and other brain areas resulted in animals gaining less weight when fed a high-calorie diet than did normal controls (23.).
Two appetite-regulating hormones produced in the digestive tract, ghrelin, an appetite stimulant and pancreatic peptide Y (PYY), a appetite suppressant have been linked to short-term feed behaviors, whereas leptin and to a lesser extent, insulin, are key to chronic weight maintenance over months and years (1). Bloom et al. reported that when ghrelin (normally produced in the stomach) was injected into human volunteers resulted in a significant increase in the amount of food they subsequently ate (24). In addition, Cummings et al. found that ghrelin levels rose an hour or two before a meal and went down to trough levels afterward (25). Bloom further reported that infusions of the PYY hormone lead to decreased eating by mice, rats and human volunteers (26) although its efficacy is controversial (27). The hormone acts in the arcuate nucleus, in this case inhibiting the activity of the appetite-stimulating neurons and stimulating the appetite-suppressive cells.
What are the options?
Diet and exercise
Many believe that a non-pharmaceutical approach is the best and most effective way to lose weight and maintain a life-long ideal body weight (1-6). This comes from the premise that if body weight is to remain stable energy intake (i.e. caloric intake) must equal energy expenditure (i.e. basal metabolism plus the effects of exercise). Thus if an individual is going to maintain an ideal body weight or is going to lose weight, sensible and well-balanced eating habits and moderate-intense exercise on a regular basis would appear to be the first set of strategies one would want to employ. And although this strategy seems straight forward, time and time again individuals fail to reach or maintain their optimal body weight for a variety of reasons which we discuss later on in this review.
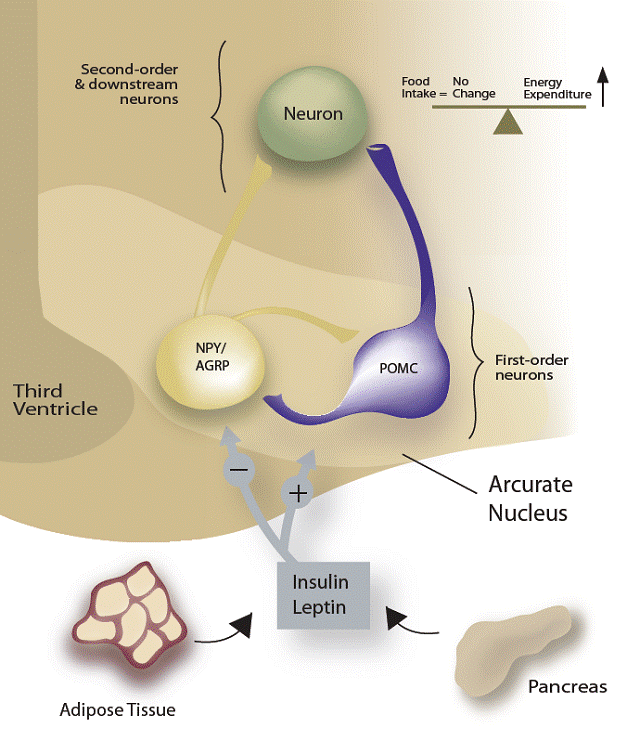 |
Figure 1. Schematic diagram of arcuate nucleus structure. Insulin and leptin in the blood stream may be stimulated by novel therapeutics. They activate the neurons POMC and NPY/AGRP resulting in increased energy expenditure. Modified from Reference #1 with permission.
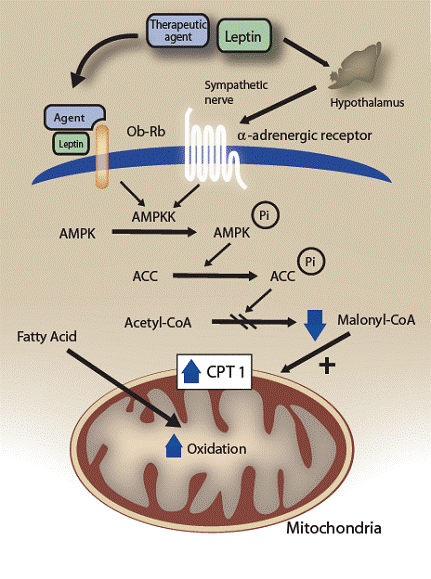
Figure 2. Potential stimulatory effect of novel therapeutics on fatty acid oxidation in muscle. The therapeutic agent could activate AMPKK/AMPK in muscle via two distinct mechanisms: one is a direct effect and the other is mediated by the hypothalamic-sympathetic nervous system. Activation of AMPK phosphorylates and inhibits ACC activity. Malonyl-CoA synthesis is inhibited, either therapeutically or through the action of leptin resulting in activation of CPT1, thereby increasing mitochondrial import and fatty acid oxidation in muscle. Modified from Reference #1 with permission.
Pharmaceuticals Currently or Previously Available for the Treatment of Obesity
Orlistat (tetrahydrolipstatin) (Trade Name: Xenical®) is a drug, which inhibits pancreatic lipase (1, 3). This prevents the gut from digesting and absorbing dietary triacylglycerols. However, lipids are not the only molecules malabsorbed; Orlistat also causes cramping and severe diarrhea in many obese patients because water molecules also fail to be taken up by the gut. Furthermore, Xenical® trials have shown that the drug helped dieting patients loses an average of 2% -3% of their body weight as compared to those dieting alone. Often patients gained the weight back after discontinuing the drug.
Sibutramine an antidepressant, hinders molecules at synapses that pick up the neurotransmitters noradrenaline and serotonin (1,3). But because the two chemical signals also control a myriad of other body processes, numerous side effects and deaths linked to the drug have been reported. In March 2002, several public citizen groups have petitioned the FDA to withdraw the drug from the market. The FDA is now conducting an investigation into these allegations (1, 3).
Phentermine one-half of the fen-phen combination (Redux) has been shown to cause heart problems and been linked to several deaths. In 1997, the “fen” component, fenfluramine, which targets the release of serotonin and has been linked to heart value disease, was banned by the FDA (1, 3). Taken together it is apparent that clinicians have few tools with which to fight obesity and weight gain; current anti-obesity drugs are not highly effective and are fraught with side effects.
Recently Jandacek and Woods have published a review on patents that are related to the treatment of obesity (3). The data generated by Jandacek and Woods (see Table 1; reprinted with permission) were obtained by searching US patents issued from January 2001 to March 2004 that included the word ‘obesity’ in the abstract (3). Among the molecular targets identified with the highest number of new patents are serotonin receptor ligands (24 patents), NPY receptor ligands (20 patents) and adrenergic receptor ligands (20 patents). Each of these molecular targets represents a different approach for the treatment of obesity.
Serotonin receptors influence most behaviors, and different serotonin receptors often have opposing actions on the same behavior. As a generality, increased serotonin tone is thought to decrease food intake and elicit weight loss (28). Because many antipsychotic drugs target the serotonin system, it is not surprising that some of these drugs have as a side effect the ability to influence appetite and body weight. Dexfenfluramine, a drug widely prescribed a decade ago to treat obesity when combined with phentermine (Fen-Phen) was in this category before being withdrawn from the market due to adverse side effects (29). An important point is that the control of mood and energy homeostasis often uses overlapping brain circuits (30). Thus, many mood-altering drugs cause changes of body weight. Some atypical antipsychotics, for example, increase body weight and thereby compromise compliance; these agents include clozapine, olanzapine, risperidone and ziprasidone (31). Several other antipsychotic or antiepileptic drugs are being used off-label to treat excess body weight, including topiramate (32) and fluoxetine (33). NPY receptor ligands are compounds based upon receptors for PYY. As previously mentioned, PYY is a peptide hormone that has been implicated in decreasing food intake in mice, rats and humans following systemic administration.
Unfortunately there are limitations of the pharmaceuticals that are currently on the market. The history of weight-loss drugs has been strewn with ones that have not been safe, from amphetamines to phentermine-fenfluramine, and their efficacy has generally been low (1, 3).
New approaches: what is in progress
The review by Jandacek and Woods (3), although an excellent overview of recent antiobesity therapeutic strategies, provides by the authors own admission, a narrow view of current research activity and omits several new areas of obesity research. Omitted areas of research include, oleoylethanolamide (OEA), a fatty acid structurally related to endogenous cannabinoids, which has been found to reduce food intake and body weight (34) and is currently under active investigation and an antagonist of the CB1 endocannibinoid receptor (Rimonabant®; Sanofi-Aventis) that reduces food intake and body weight in humans and is currently in Phase III clinical trials as a weight reduction product (35). Other potentially novel therapeutic strategies for the treatment of obesity are reviewed below.
Plant Sterols and Stanols
Plant sterols and stanols (known as phytosterols and phytostanols respectively) are naturally occurring compounds found in plants that are structurally related to cholesterol. When ingested they compete with cholesterol for absorption in the intestines, thereby reducing the amount of cholesterol absorbed into the body (36). Specifically, phytosterols and phytostanols have been shown to decrease the low-density lipoprotein (LDL) plasma cholesterol levels without any significant effects on the high-density lipoprotein (HDL) cholesterol levels in both animal and human models (36).
Table 1. US patents issued between January 2001 and March 2004 with ‘obesity’ in the abstract (Reprinted from reference 3 with permission from Elsevier*).
| Target Site and/or Treatment |
Number of Patents |
Company Assignees |
|
| Adrenomedullin receptor polypeptide |
1 |
SmithKline Beecham |
|
| Blocking alpha-2 adrenergic receptor |
2 |
Allergan |
|
| Beta-3 adrenergic receptor agonists |
18 |
BMS, Lilly, Merck, Pfizer, Warner-Lambert, Wyeth |
|
| Ciliary neurotropic factor |
3 |
Regeneron, Instituto di Richerche di Biologia Molecolare P. Angeletti |
|
| Corticotropin releasing factor (CRFP) ligands |
1 |
Neurogen |
|
| Dipeptidyl peptidase IV (DP-IV) inhibitors |
4 |
BMS, Novartis, Pfizer |
|
| Estrogen agonists/antagonists |
2 |
Pfizer |
|
| Non-absorbable fat polymers |
2 |
GelTex |
|
| Fatty acid synthase |
1 |
Bayer |
|
| Galanin receptor (GALR2) |
5 |
Merck |
|
| Glucagon-1 like peptide crystals |
2 |
Lilly |
|
| Glucagon antagonists/inverse agonists |
4 |
Novo |
|
| Glucocorticoid receptor |
4 |
Abbott, Pfizer |
|
| Glycogen synthase kinase 3 |
2 |
Chiron |
|
| GPR10 target |
1 |
Millenium |
|
| Growth hormone secretogues |
18 |
BMS, Pfizer |
|
| Leptin/Ob receptor |
8 |
Millenium, SKB |
|
| Lipase inhibitors |
7 |
Alizyme, GelTex, Laboratorie Laphal |
|
| MCH antagonists |
1 |
Schering Corporation |
|
| Melanocortin receptor agonists |
7 |
Merck, Procter & Gamble |
|
| Motilin antagonists |
1 |
Ortho McNeil |
|
| Neurokinin receptor ligands |
1 |
Neurogen |
|
| Neurotensin receptor ligand |
1 |
Pfizer |
|
| NPY, NYP1, NYP5 antagonists/modulators |
20 |
Amgen, Bayer, Hoffman-La Roche, Lilly, Merck, Neurogen, Pfizer, Schering Plough, SKB |
|
| Orexin antagonists |
1 |
SKB |
|
| Plant Stanols (FM-VP4) |
1 |
Forbes Medi-Tech Inc. |
|
| PPAR (alpha, delta, gamma) modulators |
9 |
BMS, Merck, Ortho McNeill |
|
| PTPase (PTP B) inhibitors |
10 |
Merck Frosst, Novo |
|
| Serotonin (5HT 5’ 1A’ 2C) agonists, antagonists reuptake inhibitors |
24 |
Abbott, American Home Products, BMS, Hoffman-La Roche, Pfizer, Wyeth |
|
| Thyromimetics, thyroid receptor ligands Tubby polypeptides |
8 1 |
BMS, Novartis, Pfizer SKB |
|
| Mitochondrial uncoupling proteins |
3 |
Amylin, Novartis, SKB |
|
| * Jandacek RJ, Woods SC. Pharmaceutical approaches to the treatment of obesity. Drug Discov. Today 15:9;874-880, 2004. |
|||
Our laboratory has recently completed a number of preliminary studies investigating the lipid-lowering effects of a novel phytostanol analogue FM-VP4 (disodium ascorbyl phytostanyl phosphates) in gerbils. We observed that the administration of FM-VP4 decreased body weight without altering food and water intake. After a 2-week adaptation period, adult male Mongolian gerbils (initial weight: 70-80g) were divided into six treatment groups matched for body weight and the following treatment protocol was used throughout the duration of the study: Gerbils had free access to food and water ad libitum for 8 continuous weeks (daily water and food intake were monitored and replaced), which contained either no FM-VP4 (controls), or 2.0% or 4.0% FM-VP4 incorporated into their diet (n=6 for all treatment groups) (37). Body weight and food and water intake were monitored on a weekly basis. Following 8 weeks of this regimen blood was obtained via a cardiac puncture and all the animals were sacrificed humanely. Hepatic and renal functions were assessed by measuring changes in the plasma concentration of aspartate aminotransferase (AST), alanine aminotransferase (ALT) (hepatic function) and creatinine (renal function) respectively. Animals that were administered 4% FM-VP4 in their diet demonstrated significantly lower body weight and decreased plasma TG levels following the 8 weeks of treatment compared to the other groups. Few statistical differences in food or water intake throughout the duration of the study in all treatment groups were observed (37). No significant elevations in AST, ALT and creatinine concentrations were observed in all the treatment groups compared to controls (37). Taken together, these results may be indicative of elevated energy expenditure, which could partly be explained by the alteration in pathways through which fat is metabolized. However, these results are very preliminary and further studies are warranted.
Adrenomedullin
Adrenomedullin a vasoactive peptide, which exhibits antioxidant activity and potently inhibits oxidative stress-induced vascular damage, is expressed in a number of tissues (38). Recent studies have suggested that deficiency of adrenomedullin induces insulin resistance by increasing oxidative stress (38) and that adrenomedullin and its receptor component mRNAs are concomitantly expressed in various adipose tissues, whose tissue-specific up regulation was induced during the development of obesity in rats (39). Taken together, these studies suggest that adrenomedullin may be a new member of the adipokine family and play a pathophysiological role in the development of obesity. Although its functional role has not been fully elucidated, adrenomedullin may serve as a novel therapeutic target in the future.
Peroxisome proliferator activated receptors (PPARs)
As
previously discussed, peroxisome proliferator-activated receptors (PPARs)
comprise a family of nuclear receptors that are directly linked to DNA. PPARs
are sensitive to fatty acids and cause transcriptional changes that alter
the utilization of fatty acids and glucose for energy (40). The activity of
specific PPARs has been implicated in the regulation of insulin sensitivity
and symptoms of obesity (41, 42) as well as the control of food intake (43).
Activators of PPAR ![]() as mentioned previously, including
the thiazolidinediones, increase insulin sensitivity but are associated with
weight gain through induction of adipogenesis. Activation of PPAR
as mentioned previously, including
the thiazolidinediones, increase insulin sensitivity but are associated with
weight gain through induction of adipogenesis. Activation of PPAR ![]() , however, results in increased
lipolysis and oxidation of fatty acids (44). PPAR
, however, results in increased
lipolysis and oxidation of fatty acids (44). PPAR ![]() (also called PPAR
(also called PPAR ![]() ) is implicated in the development
of diet-induced obesity (45).
) is implicated in the development
of diet-induced obesity (45).
Gastrointestinal lipase and fat metabolism inhibitors
Inhibition of pancreatic lipase limits the body of ingested calories because unhydrolyzed triacylglycerols are not absorbed from the intestine. As previously mentioned administration of Orlistat (tetrahydrolipstatin) (Trade Name: Xenical®), a pancreatic lipase inhibitor, in patients has resulted in up to a 25% decrease of fat absorption resulting in a loss of body weight. Lipase inhibiting compounds other than Xenical® have been developed, and polymers that bind fat to prevent its absorption have also been reported (3). An alternative approach to reduce absorption of ingested fat has been the development of fat that cannot be hydrolyzed. Olestra (brand name Olean®), a sucrose molecule esterified with six to eight long-chain fatty acids, has the physical properties of dietary fats but is not hydrolyzed in the intestine (46). This approach minimizes gastrointestinal side effects so long as the unabsorbed fat contains appropriate high-melting fatty acids, although gastrointestinal side effects can persist (47). The resistance to lipase also prevents hydrolysis catalyzed by bacterial lipases in the colon so that irritant fatty acids are not produced. Adding olestra to a reduced energy diet results in improved weight loss, presumably reflecting improved adherence to the diet (48).
Mitochondrial uncoupling proteins
The mitochondrial uncoupling proteins (UCP-1, UCP-2 and UCP-3) have been suggested to be involved in the development of obesity by controlling the energy expenditure. Small decreases in energy expenditure may increase the risk for human obesity (49), and genetic factors determine, at least in part, the resting metabolic rate (50). Thermogenesis is an important component of energy expenditure (EE) and body-weight maintenance. In rodents, non-shivering thermogenesis is mediated by the uncoupling protein-1 (UCP-1) in brown adipose tissue (51). UCP-1 uncouples substrate oxidation from ATP formation, thus dissipating energy as heat. However, UCP-1 is absent or very sparsely found in humans and probably does not play any role in controlling EE. With the discovery of two new members of the family of UCPs (51, 52), present in almost all human tissues, it was hypothesized that these new UCPs (UCP-2 and UCP-3) could be involved in the regulation of EE and thus obesity. Since then, numerous studies have been conducted on the regulation of these proteins and their potential importance for EE and body weight. Some studies find that obesity is associated with an increase in UCP-2 and UCP-3 mRNA in skeletal muscle (53), others find no association between obesity and UCP-3 mRNA (54), whereas others have described a negative correlation between obesity and skeletal muscle UCP-2 mRNA (55). Even fewer studies regarding the UCP mRNA expression in human adipose tissue have been performed. Oberkofler et al. (56) reported that the UCP-2 mRNA expression in intra-abdominal adipose tissue was significantly lower in obese subjects than that in lean ones; furthermore, subcutaneous adipose tissue was reported to display a non-significant lower UCP-2 mRNA expression in obese compared to that in lean individuals. Pinkney et al. (57) reported that UCP-2 gene expression in subcutaneous adipose tissue from obese individuals was significantly lower compared to that from lean ones, whereas Millet et al. (58) described a positive correlation between body mass index (BMI) and adipose tissue UCP-2 expression.
β3 adrenergic receptors and agonists
β3 -Adrenoceptor agonists are very effective thermogenic anti-obesity and insulin-sensitising agents in rodents (59). Their main sites of action are white and brown adipose tissue, and muscle. β3-Adrenoceptor mRNA levels are lower in human than in rodent adipose tissue, and adult humans have little brown adipose tissue. Nevertheless, β3-adrenoceptors are expressed in human white as well as brown adipose tissue and in skeletal muscle, and they play a role in the regulation of energy balance and glucose homeostasis. However, as Arch reports in his 2002 review, it is difficult to identify β3-adrenoceptor agonist drugs because the pharmacology of both β3 - and β1-adrenoceptors can vary; near absolute selectivity is needed to avoid β(1/2)-adrenoceptor-mediated side effects and selective agonists tend to have poor oral bioavailability (59). All weight loss is lipid and lean may actually increase, so reducing weight loss relative to energy loss. β3 -adrenoceptor agonists have a more rapid insulin-sensitising than anti-obesity effect, possibly because stimulation of lipid oxidation rapidly lowers intracellular long-chain fatty acyl CoA and diacylglycerol levels (59). This may deactivate those protein kinase C isoenzymes that inhibit insulin signaling.
DP-IV inhibitors
Dipeptidyl
peptidase IV (DP-IV or CD26) is a multifunctional glycoprotein
that contains N-terminal serine dipeptidase activity and is present
both in circulation and on the cell surface (60). DP-IV has been
implicated in pleiotropic cellular processes involving immune,
inflammatory, and endocrine functions and has been shown to cleave several
hormones and chemokines in vitro. The best validated in
vivo substrates are members of the glucagon family of peptides,
including glucagon-like peptide 1 (GLP-1), GLP-2, and glucose-dependent
insulinotropic polypeptide, which are inactivated by cleavage after
their penultimate Ala residue. GLP-1 has multifaceted actions,
including glucose-induced stimulation of insulin biosynthesis and
secretion, inhibition of glucagon secretion, regulation of gene
expression, trophic effects on ![]() cells, inhibition of food intake, and slowing
of gastric emptying. These effects contribute to the normalization
of elevated blood glucose and the control of satiety and body weight
(60). In addition, GLP-2 and glucose-dependent insulinotropic polypeptide
signaling have been implicated in the regulation of energy balance
(60). Previously, investigators have shown that upon glucose challenge,
DP-IV-/- mice have improved glucose tolerance, increased insulin,
and increased active GLP-1, consistent with the role of DP-IV in
the regulation of GLP-1. Reducing the activity of DPP-IV and thereby prolonging
the half-life and circulating levels of GLP-1 and GIP is a viable strategy
for treating obesity and improving insulin insensitivity. DPP-IV inhibitors
are also claimed to prevent degradation of NPY and PYY (61).
cells, inhibition of food intake, and slowing
of gastric emptying. These effects contribute to the normalization
of elevated blood glucose and the control of satiety and body weight
(60). In addition, GLP-2 and glucose-dependent insulinotropic polypeptide
signaling have been implicated in the regulation of energy balance
(60). Previously, investigators have shown that upon glucose challenge,
DP-IV-/- mice have improved glucose tolerance, increased insulin,
and increased active GLP-1, consistent with the role of DP-IV in
the regulation of GLP-1. Reducing the activity of DPP-IV and thereby prolonging
the half-life and circulating levels of GLP-1 and GIP is a viable strategy
for treating obesity and improving insulin insensitivity. DPP-IV inhibitors
are also claimed to prevent degradation of NPY and PYY (61).
Protein tyrosine phosphatase inhibitors
Coordinated tyrosine phosphorylation is essential for signalling pathways regulated by insulin and leptin (62). Type 2 diabetes and obesity are characterised by resistance to hormones insulin and leptin, possibly due to attenuated or diminished signalling from the receptors. Pharmacological agents capable of inhibiting the negative regulator(s) of the signalling pathways are expected to potentiate the action of insulin and leptin and therefore be beneficial for the treatment of Type 2 diabetes and obesity. A large body of data from cellular, biochemical, mouse and human genetic and chemical inhibitor studies have identified protein tyrosine phosphatase 1B (PTP1B) as a major negative regulator of both insulin and leptin signalling. In addition, evidence suggests that insulin and leptin action can be enhanced by the inhibition of PTP1B. Consequently, PTP1B has emerged as an attractive novel target for the treatment of both Type 2 diabetes and obesity. The link between PTP1B and diabetes and obesity has led to an avalanche of research dedicated to finding inhibitors of this phosphatase. With the combined use of structure and medicinal chemistry, several groups have demonstrated that it is feasible to obtain small-molecule PTP1B inhibitors with the requisite potency and selectivity. The challenge for the future will be to transform potent and selective small molecule PTP1B inhibitors into orally available drugs with desirable physicochemical properties and in vivo efficacies. PTPases influence many intracellular signaling pathways. Kennedy reported that the absence of PTP-1B in mice resulted in a resistance to obesity (63).
GPR10
GPR10, the prolactin-releasing peptide receptor, is a G protein-coupled receptor that has been presented as a target for obesity (64). GPR-10 has been identified as the receptor for prolactin-releasing peptide in the mouse hypothalamus. GPR10 knockout mice were reported to become hyperphagic and obese relative to wild type animals (65).
Non-absorbable polymers (i.e Chitosan)
An approach to the reduction of energy intake that differs from the inhibition of pancreatic lipase is that of reducing the absorption of dietary fat by binding the intestinal triacylglycerol or its hydrolysis products to non-absorbable polymers (i.e. Chitosan). This binding would reduce fat bioavailability and a source calories and energy. However, a number of studies have reported that this approach has resulted in limited (66). Mhurchu and co-workers recently reported that chitosan treatment did not result in a clinically significant loss of body weight compared with placebo in a 24-week randomised, double-blind, placebo-controlled trial in overweight and obese adults (250 participants with a body mass index of 35.5 +/- 5.1) (66).
Histamine H3 Antagonists/Inverse Agonists
A number of recent studies have suggested that histamine affects food and water consumption by acting on the central nervous system receptors (67). Hancock and colleagues reported that the blockade of postsynaptic histamine H1 receptors induced weight gain and histamine H1 receptor knockout mice displayed obese-like behaviour with increased food and water consumption (67). In contrast, antagonism of H3 receptors’, which are predominantly expressed in the CNS and act as autoreceptors in presynaptic neurons, ability to control histamine turnover has resulted in weight loss or to prevent weight gain. Currently a number of research groups are developing histamine H3 receptor antagonists as potential antiobesity agents.
Fatty Acid Hydroxylase (FAH) Inhibitors
As mentioned previously in this review stearoyl-CoA desaturase (SCD) appears to be an important metabolic control point in body weight regulation. SCD is the rate-limiting enzyme in the biosynthesis of monounsaturated fatty acids. These products are the most abundant fatty acids found in triglycerides, cholesterol esters, wax esters, and phospholipids. Inhibition of stearoyl CoA desaturase appears to induce fatty acid oxidation and weight loss and thus investigators are currently are developing inhibitors of enzymes that regulate SCD including fatty acid hydroxylase (FAH) inhibitors (68).
Johns Hopkins University (JHU), in collaboration with FASgen, is investigating fatty acid synthase (FAS) inhibitors, such as C-75, FAS-247, FAS-183, FAS-89B and FAS-57 for the potential treatment of cancer and obesity. In April 2004, JHU reported that inhibition of FAS with C-75 altered food-intake, resulting in significant weight loss in animals, with no evidence of neurotoxicity. In June 2004, preclinical data on C-75 were presented at the 64th ADA scientific sessions in Orlando, FL. Addition of C-75 (50 microM) to 3T3-L1 cells inhibited FAS activity, as did addition of cerulenin (10 microM) or triclosan (50 microM), causing incorporation of [14]C-acetate into lipids by 90, 75 and 70%, respectively. C-75 was most potent at reducing FAS and PPAR-gamma mRNA, which were reduced by up to 85% compared with control differentiated cells . In November 2004 FASgen reported that its preclinical investigations into FAS inhibitors had identified two groups of compounds with mechanisms of drug action that could work together or separately to cause weight loss. The first, including FAS-183 and FAS-57, acted on nuclei in the brain stem to curb the urge to eat; the second, including FAS-89B, stimulated the rate of fatty acid oxidation in peripheral tissues, particularly adipose tissue. FASgen noted that the compounds were effective orally and were easily absorbed from the intestine. Preclinical studies in diet-induced-obese mice had shown no toxicity or adverse reaction at doses five times higher than those required to effect weight loss.
Opinion- drugs versus life style modification
The lack of success in controlling obesity has begged the questions, as a society; are our approaches to dealing with the obesity epidemic been appropriate? Has the development of “obesity” simply been a function of “lack of awareness” that could be addressed with proper education? Or is the development of “obesity” rooted in a far deeper set of psychological issues including depression, anxiety, self-esteem or pain? Several scientific colleagues view obesity “as a form of depression in which eating is an antidepressant” (i.e. don’t need to eat excessively if you don’t exercise) (1-6), while others believe the development of obesity has occurred because of lack of education about the composition of our foods, it’s over abundant availability and the excessive commercial advertising to convince the consumer to buy more food (1-6). Yet others believe that the obesity epidemic is a result of a more evolutionary development of the sedentary life-style in our modern society. Since the beginning of the industrial revolution of the mid 1800s and technological revolution of the late 1900s there has been a dramatic change in both physical activity and food availability (5). Our society is becoming alarmingly sedentary, with some quoting that at least 70% of the United States population undertake less than 30 minutes per day of moderately intense physical activity (69). In addition, although the absolute caloric intake of an individual may actually be lower today compared with our ancestors (70) its relative intake is much higher as a function of the decreased caloric expenditure as a result of lower physical activity.
However, no matter which reason is believed to be the dominating factor, the alarming and relatively sudden development of obesity in our society is multifaceted and quite complex. A single strategy including either pharmaceutical intervention or “fad” diets may not result in a “quick-fix” answer to the problem. On the other hand, a combined pharmaceutical and behavioral modification strategy to address obesity may turn out to be the most effective approach. Individuals need to see positive results in their weight reduction program to remain motivated and if pharmaceutical intervention can supplement (but not replace) lifestyle changes to achieve their goals, this maybe a reasonable strategy. In addition, the development of pharmaceutical agents which targets specific aspects of fat metabolism and/or appetite suppression could provide useful insight into how our body processes and utilizes different components of our diet. This information alone may provide valuable information on how we as a society could optimize pharmaceutical and non-pharmaceutical strategies to address and ultimately resolve this health concern.
Acknowledgements
Canadian Institutes of Health Research University-Industry Research Chair in Drug Development to Dr. Kishor M. Wasan.
REFERENCES