Development
and Validation of Dissolution Tests for Fexofenadine Hydrochloride Capsules and
Coated Tablets
Ana R. Breier, Clésio S. Paim, Martin Steppe and
Elfrides E. S. Schapoval
Programa de Pós-Graduação em Ciências Farmacêuticas.
Faculdade de Farmácia - Universidade Federal do Rio Grande do Sul. Av.
Ipiranga, 2752. Lab. 402 Porto Alegre-RS, CEP 90610-000, Brazil
Received May
25, 2005,Revised July 4 2005, Accepted July 6, 2005, Published August 11, 2005.
pdf version
Corresponding
author: Ana R. Breier: Universidade Federal do Rio Grande do Sul, Faculdade de
Farmácia, Av.
Ipiranga, 2752, Lab 402. Porto Alegre-RS, CEP 90610-000, Brazil, E-mail: anarita_breier@hotmail.com .
ABSTRACT. Purpose: This study describes the development and validation of dissolution
tests for fexofenadine hydrochloride capsules and coated tablets using an HPLC method.
Method: The appropriate conditions
were determinate after testing sink
conditions, dissolution medium, and agitation intensity. The apparatus, paddle
and basket, were applied to tablets and capsules, respectively. Fexofenadine
hydrochloride capsules, products A and B, and coated tablets, products A, B and
C were evaluated. The best dissolution conditions tested, for the products in
each respective pharmaceutical dosage form were applied to evaluate the
dissolution profiles. The parameters of difference factor, similar factor, and
dissolution efficacy were employed. Results:
Optimal conditions to carry out the dissolution tests were 900 ml of 0.01 M hydrochloric acid as
dissolution medium, basket at 100 rotation per minute (rpm) stirring speed for
capsules and paddle at 75 rpm for tablets. The dissolution profiles for tablets
products A, B, and C and for capsules products A and B were not similar. CONCLUSION: The developed and validated
dissolution tests satisfactorily describes the time-course of the drug release.
The obtained results provided adequate dissolution profiles. The HPLC method
was validated to quantify fexofenadine capsules and coated tablets from the
dissolution tests.
INTRODUCTION
Fexofenadine,a,a - Dimethyl – 4 - [1-hydroxy – 4 - [4 - (hydroxydiphenyl-methyl) – 1
-piperidinyl] butyl]- benzene acetic acid (1) (Figure 1) is the
active carboxylic acid metabolite of terfenadine, and is a non-sedating
selective histamine H1 receptor antagonist. Unlike its precursor,
fexofenadine lacks the cardiotoxic potential, since it does not block the
potassium channel involved in repolarization of cardiac cells. Fexofenadine is effective
in the management of allergic rhinitis and chronic idiopathic urticaria for
which it is a suitable option for first-line therapy (2).
Figure 1: Chemical structure of fexofenadine
Although its distinguish importance in the
treatment of common allergic diseases, there is no monograph of this drug in
any pharmacopoeia. Moreover, the literature presents few methods related to the
quality control of fexofenadine, mainly in its pharmaceutical dosage forms.
The dissolution test has emerged as a valuable
quality control tool to assess batch-to-batch product release performance and
to assure the physiological availability of the drug (3). Its significance is based on the fact that for a drug to be
absorbed and available on the systemic circulation, it must previously be
solubilized (4).
Fexofenadine has been determined in
biological fluids by HPLC with mass spectrometry detection (5), ionspray tandem
mass spectrometry detection (6-7),
and fluorescence detection (8). The quantitation of fexofenadine in
pharmaceutical dosage forms was realized using spectrophotometric methods,
which were based in ion complex reactions (9), and HPLC methods with
ultraviolet detection (10-11). The HPLC method developed previously by our
research group (10) reported the validation of the method to quantify
fexofenadine hydrochloride capsules. There is no dissolution tests describe in
literature for fexofenadine hydrochloride in its pharmaceutical dosage forms.
This way, the aim of this work is to
present the development and validation of dissolution tests and HPLC method to
the quantitation of fexofenadine hydrochloride capsules and coated tablets in
routine quality control and from the dissolution tests, as well as to evaluate
the dissolution profiles for capsules and coated tablets.
MATERIALS AND METHODS
Instrumentation
The dissolution tests were performed in a Sotax AT7
multi-bath (n=6) dissolution test system, in accordance with the United States
Pharmacopeia (USP) general methods (12).
A
Shimadzu liquid chromatograph equipped with a model LC-10ADvp binary pump, SIL-10ADvp autosampler and model SPD-M10Avp UV detector. Detection was made at 220 nm.
SCL-10Avp system controller and CLASS-VP chromatography software were used. A
CTO-10Acvp oven was used to keep the temperature at 30 °C. The stationary
phase was a 250 x 4 mm
LiChrospherÒ 100
RP-18 octadecyl silane column (5 mm particle size) (Merck, Darmstadt, Germany).
The mobile phase was prepared by mixing 50 mM ammonium acetate buffer and acetonitrile (50:50, v/v) – pH 3.2 (adjusted with hydrochloric acid 0.1N). The injection volume was 20 ml and
the run time was 10 minutes. The mobile phase was filtered using a 0.45 mm
membrane filter (Milipore, Milford,
MA) and degassed with helium. The
mobile phase flow rate was 1.0 ml.min-1.
Materials and Reagents
Fexofenadine hydrochloride
reference substance (99.6%) was obtained from Aventis Pharma (São Paulo, Brazil),
whereas the pharmaceutical formulations containing fexofenadine hydrochloride
were obtained commercially.
Analytical
reagent grade chemicals were used. Buffer solutions pH 1.2, pH 4.0, and pH 6.8
were prepared according USP 28 (12).
Fexofenadine capsules:
Product A- labeled to contain 60 mg of the drug and the
following excipients: pregelatinized starch, lactose, croscarmellose sodium,
and microcrystaline cellulose. This product is the reference brand in Brazil.
Product B- labeled to contain 60 mg of the drug and the
following excipients: lactose, croscarmellose sodium, and microcrystaline
cellulose.
Fexofenadine coated tablets:
Product A- labeled to contain 120 mg of the drug and the
following excipients: pregelatinized starch, lactose, croscarmellose sodium,
and microcrystaline cellulose. This product is the reference brand in Brazil.
Product B- labeled to contain 120 mg of the drug and the
following excipients: lactose, croscarmellose sodium, and microcrystaline
cellulose;
Product C- labeled to contain 120 mg of the drug and the
following excipients: lactose, croscarmellose sodium, and microcrystaline
cellulose.
Dissolution tests conditions
Fexofenadine sink
conditions were determined in different solvents. The solubility of the drug
was tested using an amount of fexofenadine hydrochloride equivalent a three
times of the dose in the pharmaceutical formulation in 900 ml of HCl 0.1 M, HCl 0.01 M, phosphate buffer pH
1.2, pH 4.0 and phosphate buffer pH 6.8. The solubility in water was not
tested, since it is not an ideal dissolution medium. Then, three dissolution
medium were chosen to be tested in the drug release percent – 0.1 M hydrochloric acid, 0.01 M hydrochloric acid,
and phosphate buffer pH 6.8. Thus, stirring speeds of 75 rpm and 100 rpm for
capsules and 50 rpm and 75 rpm for tablets were tested. For dissolution tests,
900 ml of each medium were deaerated in
ultrasonic bath for 15 minutes and maintained at 37 ± 0.5 °C and USP apparatus, paddle or
basket, were used for tablets and capsules, respectively. The test time was set
on 60 min (13).
HPLC
Preparation of standard
solutions
The standard solution was
prepared using an amount of powder equivalent to 10 mg of fexofenadine
hydrochloride that was transferred to a 50 ml volumetric flask with mobile
phase (0.2 mg ml-1). Aliquot of 4 ml of this standard
solution were transferred to 20 ml volumetric flask and diluted with the same
diluent obtaining the final concentration of 40.0 mg ml-1. The solution was filtered in a 0.45 mm membrane filter before the
injection in the column.
Preparation of sample solutions
The sample solutions were
prepared using amounts of powder equivalent to 10 mg of fexofenadine
hydrochloride tablets which were transferred to 50 ml volumetric flask with
mobile phase (0.2
mg ml-1). These solutions were
kept in the ultrasonic bath for 15 minutes and shaken for 15 minutes. Aliquots
of 4 ml of the solutions were transferred to 20 ml volumetric flasks and
diluted with the same diluent obtaining the final concentration of 40.0 mg ml-1. The solutions were filtered in a 0.45 mm membrane filter before the
injection in the column.
Dissolution tests and HPLC validation
The dissolution tests were
validated to fexofenadine hydrochloride capsules and tablets through the
determination of specificity, linearity, intermediate precision, and solutions
stability (13-14).
In our previous work
(10), the HPLC method was developed and validated for the quantitation of
fexofenadine hydrochloride capsules. In this work, in order to validate the
HPLC method for coated tablets, the parameters of specificity, linearity,
precision, accuracy and robustness were evaluated.
Specificity: the dissolution tests specificity was evaluated
by preparing samples of each placebo of the commercial formulation of capsule
and tablets (cited in Section 2.2). These samples were transferred to separate
vessels with 900 ml of the dissolution medium and stirred for 1 h at 150 rpm
using the respective method apparatus. The interference of the excipients of
each formulation was evaluated by UV and HPLC. The evaluation of the HPLC
method specificity was performed by preparing placebo tablets containing the
same excipients of the commercial products (cited in Section 2.2). The
solutions were prepared using the same procedure described for the sample
solutions (Section 2.4.2) and injected three times.
Linearity: In order to assess the
linearity of the method, seven doses of the reference substance (20.0; 30.0;
40.0; 50.0; 60.0; 70.0; and 80.0 mg ml-1) were used
at HPLC method for the standard curves. The calculation of regression line was
employed by the method of least squares.
Precision: The evaluation of the intermediate precision of
the dissolution tests was performed using a well-characterized lot of the drug
product of tight content uniformity and compared with the results of the
dissolution tests. According USP 28 (12), the content uniformity was evaluated assaying
ten capsules or tablets individually and calculating the content of
fexofenadine hydrochloride of each one. For the HPLC method, the repeatability (intra-assay) and
intermediate precision (inter-assay) were determined by assaying samples of
coated tablets, at the same concentration (40.0 mg ml-1), under the
same experimental conditions described in 2.4.2 section, during the same day
and in three different days, respectively. The intermediate precision
(inter-assay) was evaluated by comparing the assays on these three different
days. The relative standard deviation (RSD) was determined.
Accuracy: This parameter was determined by the recovery test,
which consists in adding known amounts of fexofenadine reference substance to
the samples. Aliquots of 2.5, 5.0, and 7.5 ml of a 0.1 mg ml-1
fexofenadine hydrochloride standard solution (10.0 mg, 20.0 mg and 30.0 mg, respectively, corresponding to 25.0, 50.0 and 75.0%
of the sample concentration) were added to three commercial samples solutions,
respectively, prepared as cited in Section 2.4.2. Each solution was prepared in
triplicate and each one was injected in triplicate.
Robustness: The robustness was tested by changing the
following parameters of the HPLC method (one by one): mobile phase proportion –
it was used 50 mM ammonium acetate buffer and acetonitrile (45:55, v/v –
pH 3.2) mobile phase; mobile phase pH – it was used pH 2.6 and pH 4.8; stationary phase – it was used a MetaSil
octadecyl silane (250 x 4.6
mm, 5 mm – MetaChem Technologies, Torrance,
USA); and another
liquid chromatograph – the quantitation was performed in a Shimadzu liquid chromatograph
equipped with a model LC-10AS pump, Rheodyne
injector with a 20 ml loop and model SD-10A UV detector.
Solutions stability: the solutions stability was analyzed over a
specified period of time, verifying the response of the sample solution stored
at room temperature.
Dissolution profiles
The dissolution profiles were obtained after the
determination of the best dissolution conditions tests. Aliquots of 15 ml were
withdrawn of each vessel and the same volume of the dissolution medium was
replaced to maintain a constant total volume. The times selected were 5; 10;
15; 30; 45; and 60 minutes. Twelve samples were assayed for each dissolution
profile. The withdrawn samples were filtered in 0.45 mm and diluted with mobile
phase to 40 mg ml-1 to HPLC
quantitation.
Release dissolution profiles comparison
The dissolution profiles were compared through the
calculation of dissolution efficiency (DE) and model-independent simple method.
The DE was calculated from the area under the dissolution curve at time ti (measured using the
trapezoidal rule) and expressed as a percentage of the area of the rectangle
described by 100% dissolution in the same time.
The model-independent simple
method includes the difference factor (f1)
and the similarity factor (f2).
The f1 factor
measures the percent error between two curves over all time points. The percent
error is zero when the test and drug reference profiles are identical and
increase proportionally with the dissimilarity between the two dissolution
profiles.
The f2 factor is a logarithmic transformation of the
sum-squared error of differences between the test and the reference products
over all time points. This factor is 100 when the test and reference profiles
are identical and tends to 0 as the dissimilarity increases. Two dissolution
profiles are declared similar if f1
is between 0 and 15 and if f2
is between 50 and 100 (15-16).
RESULTS AND DISCUSSION
The sink
conditions tested showed that fexofenadine hydrochloride bulk was soluble in
HCl 0.1 M,
HCl 0.01 M
and phosphate buffer pH 6.8. Then, dissolution tests for fexofenadine
hydrochloride tablets product A were performed using these three dissolution
medium at the stirring speed of 75 rpm, to investigate the drug release in each
medium (Figure 2). The results show that HCl 0.01 M was the best
dissolution medium, since it provides highest drug release percent.
For capsules, the basket
method is routinely used at an agitation speed of 50 to 100 rpm. For tablets,
the paddle method is frequently used at 50 or 75 rpm (14). Thus, stirring
speeds of 75 rpm and 100 rpm for capsules products A and B (Figure 3) and 50
rpm and 75 rpm for coated tablets product A (Figure 4) were tested. The
statistical t-student test at 0.05
significance level was applied to compare the drug release percent (DR%), using
75 or 100 rpm for capsules (Table 1) and 50 or 75 rpm for tablets (Table 2).
The P-values presented for capsules were greater than the delineated
significance level, indicating that there was no statistically significant
difference between the drug release percent and suggested that any of the
stirring speed could be used, for products A and B. However, it was observed that
stirring speed of 100 rpm presents high drug release percent until 30 minutes.
The P-value for tablets was smaller than the delineated significance level,
indicating that there is statistically significant difference between the drug
release percent and suggested that the stirring speed of 75 rpm is better than
50 rpm. Thus, the stirring speed of 100 rpm for capsules and 75 rpm for tablets
were chosen.
The reversed-phase liquid chromatography
method was developed and validated for fexofenadine hydrochloride in coated
tablets. The validation analytical parameters described in the guidelines (12,
17) were evaluated. The type of method and its respective use determine which
parameters should be evaluated. It is the responsibility of the analyst to
select the parameters considered relevant for each method (18).
The specificity of the dissolution test was evaluated through
the analysis of placebo capsules and tablets from a dissolution test using the
HPLC and UV methods. The analysis by UV shows that the excipients from capsules
and coated tablets absorbed at 220 nm (Figure 5), which characterize
interference in the analysis. So, the UV method can not be use to quantify
fexofenadine hydrochloride capsules and coated tablets from the dissolution
tests.
The specificity test by HPLC
demonstrated that the excipients from capsules and tablets do not interfere in
the drug peak (Figure 6). Thus, the HPLC method is useful to quantify
fexofenadine hydrochloride in pharmaceutical formulation from the dissolution
tests. The chromatogram obtained through the injection of the placebo solution
did not present any other peak in the same retention time (5 minutes) of
fexofenadine hydrochloride (Figure 7). The chromatographic peak purity tool was
used in order to verify the purity. This tool works analyzing the peak and
given a value between 0 and 1. The obtained value was 0.9999, this result shows
that the analyzed peak was only fexofenadine hydrochloride, without
interference.
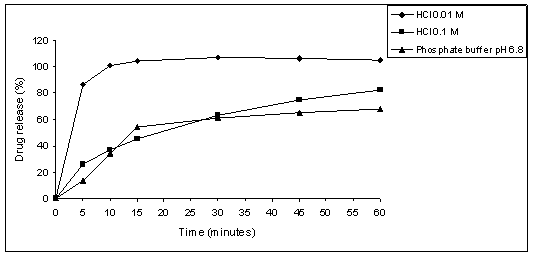
Figure 2. Product A - coated tablets dissolution
profiles using HCl 0.01 M,
HCl 0.1 M
or phosphate buffer pH 6.8 as dissolution medium and paddle at 75 rpm.
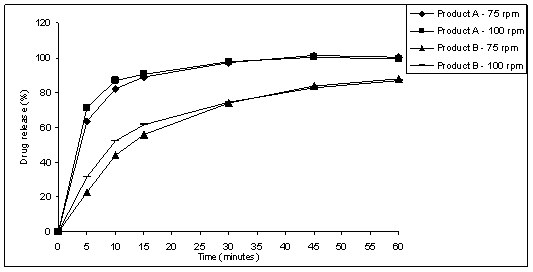
Figure 3: Products A and B - capsules
dissolution profiles using HCl 0.01 M, and basket at 75 rpm or 100 rpm.
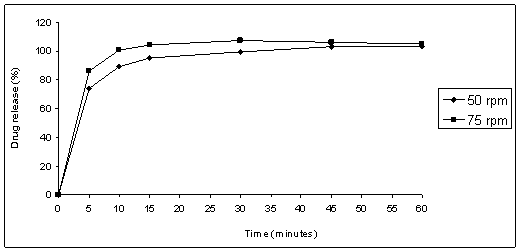
Figure 4: Product A - coated tablets
dissolution profiles using HCl 0.01 M, and paddle at 50 rpm or 75 rpm.
Thus, it was proved that the
peak at 5.0 min was not suffering interference of any
excipients from the formulation.
To assess the linearity, three
standard curves for fexofenadine hydrochloride were constructed, plotting
concentrations (mg ml-1) versus
absolute area (mV s) and showed good linearity on the 20.0-80.0 mg ml-1 range. The representative linear
equation was y = 31260.64 x + 31492.44, where x is concentration and y is the peak absolute area. The
correlation coefficient was r = 0.9999, indicating good linearity. The data
were validated by means of the analysis of variance, which demonstrated
significative linear regression and no significant linearity deviation (p< 0.05) (19).
Table 1: Products A and B capsules
dissolution tests results (n=12), using different stirring speeds and HCl 0.01M
as dissolution medium.
|
Product
|
min
|
DR%
|
t-test
|
p
|
|
75
rpm
|
100
rpm
|
|
A
|
0
5
10
15
30
45
60
|
0
63.85
82.32
89.17
97.50
101.19
100.43
|
0
71.40
87.14
90.74
97.82
100.57
99.29
|
1.468
|
0.19
|
|
B
|
0
5
10
15
30
45
60
|
0
22.92
44.11
56.04
73.77
83.69
87.94
|
0
31.38
52.15
61.40
74.54
82.68
86.73
|
1.814
|
0.12
|
The
intermediate precision of the dissolution tests was verified through the
comparison of the results of uniformity of content and the percentage drug
release. The mean values found for the uniformity of content of product A and B
capsules were 111.61% (RSD = 1.80) and 101.20% (RSD = 2.41), respectively. The
drug release percent were 99.29%, for product A and 86.73% for product B. The
difference between the uniformity of content and drug release percent can be
explained by the incomplete dissolution of gelatin wrapping, which kept an
amount of the drug inside. However, this effect do not interfere the
dissolution test, because more than 70% of drug was dissolved in 30 minutes in
all tests.
Table 2: Product A coated tablets
dissolution tests results (n=12), using different stirring speeds and HCl 0.01M
as dissolution medium.
|
Product
|
min
|
DR%
|
t-test
|
p
|
|
50
rpm
|
75
rpm
|
|
A
|
0
5
10
15
30
45
60
|
0
74.12
89.25
95.36
99.55
103.10
103.01
|
0
86.28
100.59
103.97
107.29
106.12
105.05
|
3.57
|
0.012
|
The mean
values found for uniformity of content to product A, product B, and product C
coated tablets were 107.01% (RSD=0.51), 103.03% (RSD=1.11), and 103.87%
(RSD=1.2), respectively. The drug release percent were 105.05%, 103.85%, and
105.06%, for products A, B, and C, respectively. In all tests, almost all drug
was dissolved. These results show the good precision of the dissolution tests.
The experimental values
obtained for the determination of fexofenadine hydrochloride in samples are
presented in Table 3. The low relative standard deviation (RSD) of 1.67
(intra-day precision), and 0.12 (inter-day precision) showed the good precision
of the method.
The accuracy expresses the agreement between the
accepted value and the value found. The mean recovery was found to be 99.94%
for the coated tablets (Table 4). This value shows the good accuracy of the proposed
method.
The
robustness of the method evaluated by changing the mobile phase proportion, 50 mM ammonium acetate buffer and acetonitrile (45:55, v/v; pH,
3.2) demonstrated an increase on the
retention time of the drug. The use of pH 2.6 resulted in a decrease in
the retention time. The method was robust with these two modifications. When pH
4.8 was used, the retention time was about 4.3 min, but the peak become wide,
probably because in this pH the drug is in the ionizated form. The effect of using MetaSil octadecyl silane (250
x 4.6 mm,
5 mm) as
stationary phase has increased the retention time in two minutes. Even so, the
method was robust.

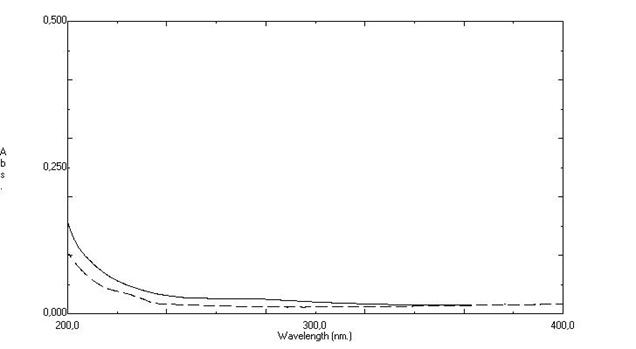
Figure 5: Absorbance vs wavelength specificity
of fexofenadine hydrochloride capsules and coated tablets from the dissolution
test by UV.
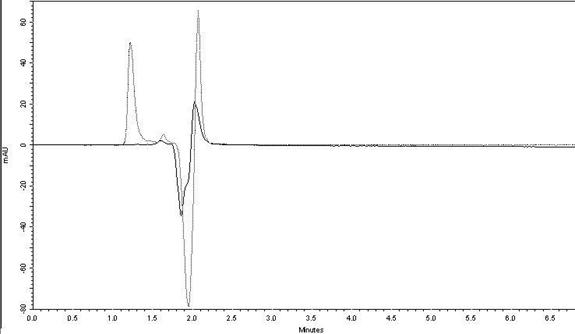
Figure 6: Specificity test for
fexofenadine hydrochloride capsules and coated tablets from the dissolution
test by HPLC.
The
last experiment was the quantitation in another liquid chromatograph (Shimadzu equipped with a model
LC-10AS
pump, Rheodyne injector
with a 20 ml loop and model SD-10A UV detector) where the retention time was a little high (about 5.4
minutes), but it was possible to quantify the drug satisfactorily, confirming
the robustness of the method. At that rate, it was possible to demonstrate that
the developed method was robust with all the changes employed, except for the
use of pH 4.8 in
the mobile phase.
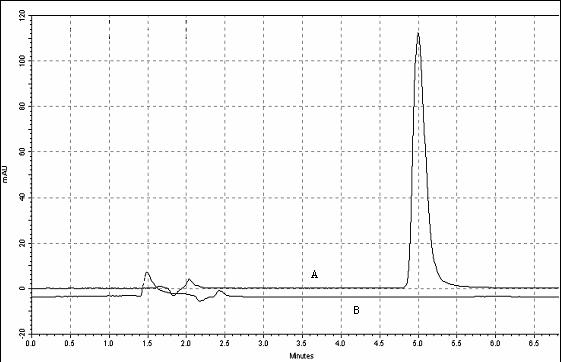
Figure 7: Chromatograms of fexofenadine
hydrochloride coated tablets sample solution 40
mg ml-1 (A) and
placebo solution (B). Chromatography conditions: acetonitrile, 50 mM ammonium
acetate buffer (50:50, v/v) at pH 3.2 mobile phase; flow rate of 1,0 ml ml-1;
Lichrospherâ 100 RP-18 (250
´ 4.0
mm, 5
mm) stationary phase;
ultraviolet detection at 220 nm; temperature
of 30 °C;
injection volume of 20
ml.
Table
3: Experimental values of fexofenadine hydrochloride coated tablets
obtained in commercially available sample, using the HPLC method.
|
Sample
|
Precision, Intra-assay
|
|
1 st day
|
2 nd day
|
3 th day
|
|
1
|
97.86
|
100.02
|
99.24
|
|
2
|
100.83
|
99.35
|
99.12
|
|
3
|
96.91
|
98.58
|
98.83
|
|
4
|
100.09
|
99.21
|
99.79
|
|
5
|
100.66
|
99.28
|
99.92
|
|
6
|
98.14
|
98.01
|
99.01
|
|
Mean
|
99.08
|
99.24
|
99.32
|
|
RSD
|
1.67
|
0.48
|
0.44
|
|
Precision
Inter-assay
|
99.21
|
|
RSD
|
0.12
|
The
stability test of the solutions shows that fexofenadine hydrochloride was
stable in HCl 0.01 M
at least 24 hours at room temperature and this way it can be analyzed with
precision during the dissolution assay.
The comparison of the
dissolution profiles for the different products cited in section 2.2 was
realized. The results of dissolution efficiency (DE), difference factor (f1) and the similarity factor
(f2) are presented in
Tables 5 and 6 for coated tablets and capsules, respectively. Since product A
is the reference brand, the factors f1
and f2 were calculated
between product A and B for tablets and capsules. Two dissolution profiles are
declared similar if f1 is
between 0 and 15 and if f2
is between 50 and 100. The results of f1
and f2, 36.23
and 17.45, respectively, for the comparison of product A and B, showed that the
profiles are not similar. For product C (coated tablets) these factors were not
calculated, because the dissolution was very fast (more than 85% in 15
minutes). The dissolution efficiency was calculated for all products capsules
and tablets. The analysis of variance of the DE values shows that the profiles
are not similar for tablets and capsules.
Table 4: Experimental values obtained
in the recovery test for fexofenadine hydrochloride coated tablets, using the
HPLC method.
|
Sample
|
Amount of reference (mg)
|
Recovery*
%
|
Mean
|
RSD
|
|
Added
|
Recovered
|
|
R1
|
0.25
|
0.251
|
100.35
|
99.94
|
1.67
|
|
R2
|
0.50
|
0.491
|
98.10
|
|
R3
|
0.75
|
0.760
|
101.36
|
Typical acceptance criteria
for the amount of drug dissolved are in the range of 75% to 80% dissolved. Acceptance criteria including test times
are usually established on the basis of an
evaluation of the dissolution profile data (14). In this article, it was
observed that for all products a dissolution of 70% /30 min. So, this
acceptance criterion was utilized.
Table 5:
Comparison of coated tablets dissolution profiles through the dissolution
efficiency (DE), difference factor (f1)
and the similarity factor (f2).
|
Parameter
|
Product A
(reference)
|
Product B
|
Product C
|
|
DE
|
103.85
|
101.64
|
78.85
|
|
f1*
|
36.23
|
-
|
|
f2*
|
17.45
|
-
|
|
* calculated
between products A and B.
|
Table 6: Comparison of capsules
dissolution profiles through the dissolution efficiency (DE), difference factor
(f1) and the similarity
factor (f2).
|
Parameter
|
Product A
(reference)
|
Product B
|
|
DE
|
95.19
|
73.07
|
|
f1*
|
28.90
|
|
f2*
|
27.62
|
|
*calculated between
products A and B.
|
CONCLUSIONS
In this work, it was developed
and validated dissolution tests and evaluated dissolution profiles for
fexofenadine hydrochloride in capsules and coated tablets. The use of 900 ml of
0.01 M
hydrochloric acid at 37
°C, basket at the stirring speed of 100 rpm and paddle
at the stirring speed of 75 rpm as apparatus for capsules and coated tablets,
respectively, and 60 min of test provided satisfactory results for all
products. The comparison of the obtained dissolution profiles was realized by
DE and the factors f1 and f2 and show that the profiles
were not similar neither for capsules products A and B, nor for tablets
products A, B and C. However, for all products the drug delivery was
satisfactory, since at least 70% was dissolved in 30 minutes. The HPLC method was
validated to the routine quality control of fexofenadine hydrochloride in coated
tablets and was satisfactory in the quantitation of fexofenadine hydrochloride
capsules and coated tablets from the dissolution tests. The UV method could not
be used, since it lacks in specificity.
ACKNOWLEDGEMENTS
The
authors thank to CAPES and the Brazilian Pharmacopoeia
by the financial support and LEPCQ.
REFERENCES
1. The Merck Index, an
Encyclopedia of Chemicals, Drugs, and Biologicals. Merck & CO., INC.
Whitehouse Station, NJ, 2001.
2. Simpson, K. and Jarvis, B., Fexofenadine: a review of its use in
the management of seasonal allergic rhinitis and chronic idiopathic urticaria. Drugs, 59:301-321, 2000.
3. Shah, V.P., Noory, A., Noory, C., McCullough, B., Clarke, S.,
Everett, R., Naviasky, H., Srinivasan, B.N., Fortman, D., and Skelly, J.P., In
vitro dissolution of sparingly water-soluble drug dosage forms. Int J Pharm, 125:99-106, 1995.
4. Furlanetto,
S., Maestrelli, F., Orlandini, S., Pinzauti, S., and Mura, P., Optimization of
dissolution test precision for a ketoprofen oral extended-release product. J Pharm Biomed Anal, 32:159-165, 2003.
5. Hofmann, U., Seiler, M.,
Drescher, S., and Fromm. M.F., Determination of fexofenadine in human plasma
and urine by liquid chromatography-mass spectrometry. J Chrom B, 766:227-233, 2002.
6. Gergov, M., Robson,
J.N., Ojanperä, I., Heinonen, O.P., and Vuori, E., Simultaneous screening and
quantitation of 18 antihistamine drugs in blood by liquid chromatography
ionspray tandem mass spectrometry. Forensic
Sci Int, 121:108-115, 2001.
7. Fu, I., Woolf, E.J., and Matuszewski, B.K., Determination of
fexofenadine in human plasma using 96-well solid phase extraction and HPLC with
tandem mass spectrometric detection. J
Pharm Biomed Anal, 35: 837-846, 2004.
8. Uno,
T., Yasui-Furukori, N., Takahata, T., Sugawara, K., and
Tateishi, T., Liquid chromatographic determination of fexofenadine in human
plasma with fluorescence detection. J
Pharm Biomed Anal, 35:937-942, 2004.
9. Gazy, A.A., Mahgoub, H.,
El-Yazbi, F.A., El-Sayed, M.A., and Youssef, R.M., Determination of some
histamine H1-receptor antagonists in dosage forms. J Pharm Biomed Anal, 30:859-867, 2002.
10. Breier, A.R., Menegola, J., Paim, C.S., Steppe,
M., and Schapoval, E.E.S., Development and validation of a liquid
chromatographic method for fexofenadine hydrochloride in capsules. J of AOAC Int, 87: 1093-1097, 2004.
11. Radhakrishna, T., and
Reddy, G.O. Simultaneous determination of fexofenadine and its related
compounds by HPLC. J Pharm Biomed Anal,
24:755-767, 2002.
12. United States
Pharmacopoeia, 28th.ed. Rockville, United States Pharmacopoeial Convention,
2005.
13. Marques, M.R.C., and
Brown, W., Desenvolvimento e validação de métodos de dissolução para formas
farmacêuticas sólidas orais. Revista
Analytica, 01:48-51, 2002.
14. Pharmacopeial Forum.
Pharmacopeial Previews, 30; (1) [Jan.-Feb. 2004].
15. Costa, P., and Lobo,
J.M.S., Modeling and comparison of dissolution profiles. Eur J Pharm Sci, 13:123-133, 2001.
16. Brasil, Resolução-RE n°
310, de 1° de setembro de 2004. Anvisa, 2004.
17. International
Conference on Harmonization of
Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), Guideline on Validation of Analytical
Procedure-Methodology, 1996.
18. Ermer,
J., Validation in pharmaceutical analysis, part I, an integrated approach. J Pharm Biomed Anal, 24: 755-767, 2001.
19. Farmacopéia Brasileira, 4th Ed, Ahteneu, Rio de Janeiro, 1988.