J Pharm Pharmaceut Sci (www.cspscanada.org) 8(1):47-53, 2005
Steady-state bioequivalence study of clozapine tablet in schizophrenic patients.
1Wongwiwat Tassaneeyakul*, 2Khanogwan Kittiwattanagul, 3Suda Vannaprasaht, 4Jureeporn Kampan, 3Arporn Tawalee, 3Prapawadee Puapairoj, 3Siriporn Tiamkao, 2Srisaard Juthagridsada, 3Veerapol Kukongviriyapan, 3Wichittra Tassaneeyakul
1Department of Toxicology, Faculty of Pharmaceutical Sciences, Khon Kaen University, Khon Kaen, Thailand; 3Department of Pharmacology and 4Department of Pathology, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand; 2Khon Kaen Rajanagarindra Psychiatric Hospital; Khon Kaen, ThailandReceived 16 December 2004, Revised 20 January, 2005, Accepted 28 January 2005, Published 25 February 2005
PDF Version
Abstract
PURPOSE: To compare the bioavailability of two clozapine formulations (100 mg Clozaril tablet from Novartis Pharmaceuticals UK Ltd., UK, as a Reference formulation and 100 mg Cloril tablet from Atlantic Laboratories Corp., Ltd., Thailand, as a Test formulation). The present study was conducted under real-life conditions in schizophrenic patients using a steady-state, multiple-dose, randomized crossover design to avoid the risk of adverse effects in healthy volunteers and pharmacokinetic difference between single and multiple-dose of the drug. METHODS: The subjects received 100 mg bid of either the Reference formulation or the Test formulation for 7 days. At day- 7 of each study phase, blood samples were collected at 0, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10 and 12 h after drug administration. Plasma was separated and stored at -80°C until assay. The plasma concentration of clozapine was determined by high performance liquid chromatography. Pharmacokinetic parameters were calculated from the observed plasma-concentration time profiles. The bioequivalence between the two formulations was assessed by calculating individual peak plasma concentrations (Cmax) and area under the concentration-time curve (AUC0-12 h) ratios. RESULTS: All subjects well tolerated both clozapine formulations. No serious side effects were reported. The Tmax, terminal half-life and the total plasma clearance of clozapine (uncorrected for bioavailability) observed in the present study were comparable to those observed in other previous reports. All of the pharmacokinetic parameters investigated in the present study calculated from the subjects after administration of Test and Reference formulations were close. The 90% confident interval for the ratio of means for the lnCmax (0.9784-1.0622) and lnAUC0-12h (0.9559-1.0441) are within the guideline range of bioequivalence (0.80 to 1.25). CONCLUSION: The result demonstrated that the Test formulation was bioequivalent to the Reference formulation (Clozaril) when orally administered in schizophrenic patients, in terms of both the rate and extent of absorption.
Introduction
Clozapine, a dibenzodiazepine derivative, is an atypical antipsychotic drug with potent antipsychotic properties. Unlike other conventional neuroleptic drugs, clozapine does not appear to produce significant extrapyramidal syndrome and tardive dyskinesia (1, 2). This drug is indicated for treatment of severely ill schizophrenic patients who fail to respond to conventional antipsychotic drug such as chlorpromazine or haloperidol (3, 4). Clozapine acts as an antagonist of dopamine receptors in the mesolimbic system (1, 5). The binding ratio of clozapine to serotonine (5-HT2A) receptor and dopamine (D2) receptor is higher than other conventional antipsychotic drugs (6, 7). This drug also shows higher affinity to dopamine D1 and D4 receptors as well as acts an antagonist at adrenergic, cholinergic, histaminergic and seronergic receptors (5, 8).
Clozapine is rapidly and almost completely absorbed following oral administration with Tmax of 1.5-2 h and the maximum effect of the drug appears approximately 4 h after administration (5, 8, 9). Approximately 95% of the drug binds to plasma proteins and about 50% undergoes first-pass metabolism before being distributed to the systemic circulation (10, 11). This drug is extensively metabolized by cytochromes P450 and the major metabolites detected in urine are N-oxide, N-desmethyl and hydroxylated derivatives (9, 12, 13). The desmethyl metabolite has only limited pharmacological activity while the hydroxylated and N-oxide derivatives are inactive. Only trace amounts of the parent compound is found in urine and feces.
Since clozapine is commonly used for treatment of schizophrenia, several generic products are now available. In order to assure the therapeutic equivalence of these generic products, the bioequivalence study needs to be investigated. Previous evidence from bioequivalence studies revealed that administration of clozapine to healthy volunteers may result in serious adverse effects such as hypotension, bradycardia, syncope and asystole (14). The risk of agranulocytosis or cardiomyopathy has also been reported in some patients (15, 16) which make healthy volunteers appear inappropriate for pharmacokinetic study. Moreover, differences in the pharmacokinetic parameters between the single- and multiple-dose of clozapine have been documented. The mean AUC, volume of distribution and half-life were significantly increased in the multiple-dose study, which implied additional pharmacokinetic compartments, larger volume of distribution or concentration dependent pharmacokinetics during multiple dose administration (17). Therefore, the present study was carried out to compare the pharmacokinetic profiles and evaluate the bioequivalence of the two clozapine formulations in 18 schizophrenic patients under real-life conditions. This should avoid the risk of unnecessary adverse events and any discrepancy in the pharmacokinetics of clozapine between the single- and multiple-dose. The Test clozapine tablet (100 mg) formulation from Thailand was compared with the Reference clozapine tablet (100 mg) formulation produced by Novartis Pharmaceuticals UK Ltd., Horsham, UK, using a steady-state, multiple-dose, randomized crossover study.
Materials and Methods
Drugs and Chemicals
Standard clozapine was purchased from Council of European Pharmacopoeia, Strasbourg, France. Protriptyline HCl was obtained from USP Inc., Rockville, MD, USA. Other chemicals used in the study were of analytical grade and purchased from BDH Chemicals Ltd, Poole, England. Clozapine formulations employed in this study were 100 mg Clozaril tablet (Novartis Pharmaceuticals UK Ltd., Horsham, UK, Lot No. U43536, manufacturing date 12/2000, expiry date 12/2003), and 100 mg Cloril tablet (Atlantic Laboratories Corp., Ltd., Bangkok, Thailand, Lot No. PD020057, manufacturing date 17/8/2002, expiration date 17/8/2004).
Subjects
Eighteen male Thai schizophrenic patients enrolled in the study ranging in age from 18 to 55 years (mean ± SD, 35.8 ± 9.9). These subjects were regularly given clozapine 100 mg bid in order to control of their symptoms. Their body mass indices were in the range of 20-25. Each subject was physically healthy as indicated by their medical histories, physical examinations and standard clinical laboratory tests. Exclusion criteria included those who had a medical history of liver disease, kidney disease, cardiovascular diseases, blood disorders, gastrointestinal disorders, hepatitis, drug abuse, alcoholism, AIDS or HIV sero-positive. Subjects with an allergy to clozapine or other anti-psychotic drugs such as bezodiazepines or atypical anti-psychotics were excluded. The study protocol was approved by the Thai-Food and Drug Administration and Khon Kaen University Ethics Committee for Human Research, Khon Kaen, Thailand. All subjects were informed, both verbally and in writing, about the experimental procedures and the purposes of the study. Written informed consent was obtained for their participation in the study.
Study Design
The study was conducted using a multiple-dose, randomized, two-way crossover design. The subjects randomly received either 100 mg twice daily of the clozapine formulation [i.e. Reference formulation, Clozaril (Novartis Pharmaceuticals UK Ltd., Horsham, UK) or Test formulation, Cloril (Atlantic Laboratories Corp., Ltd., Bangkok, Thailand)] after breakfast (8.00 AM) and after dinner (8.00 PM) for 7 days. Drug administration was performed by the staff nurses of the Rehabilitation Center. The timing of drug administration was recorded and tablet counts were rechecked by researchers to ensure the subject's compliance. On day- 5 and day-6 of each study phase, blood samples were collected before the morning dose administration of the drug in order to check whether or not the blood level of clozapine has reached steady state. On day-7 of each study phase, subjects received breakfast at 6.00 AM. At approximately 8.00 AM pre-dose blood samples were drawn from the subjects through an indwelling catheter placed in the arm. The subjects were subsequently given one tablet of 100 mg clozapine orally with 200 ml water and blood samples were collected at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10 and 12 h after drug administration (via IV catheter). In day-8 to day-14 of the study, subjects received the same treatment as the first 7 days of the study except for a different clozapine formulation. Blood samples were centrifuged and plasma were separated, transferred to new tubes, and stored at -80°C until assay.
During the study period, all subjects received close attention from the doctor and nurses. All meals provided for the subjects particularly the meal on day-7 and day-14 in which the pharmacokinetic study was conducted was similar. Soft breakfast was provided 2 h before drug administration. Lunch and dinner was provided at 4 and 10 h, respectively, after drug administration. During meals, subjects were allowed to drink water and had other normal activities.
Determination of Clozapine in plasma
Plasma concentrations of clozapine were assayed by high performance liquid chromatography using the modified method of Chung et al., 1993 (18) as follows: 0.5 ml of plasma sample was transferred in to the screwed capped tube. 40 ml of an internal standard (protriptyline 31.2 mg/ml) and 200 ml of 2 M NaOH were added in to the plasma tube. The sample was then thoroughly mixed and left at room temperature for 5 min. After that, 5 ml of hexane: isoamyl alcohol (98.5:1.5 v/v) was added and vortex mixed at full speed for 2 min. The organic and aqueous phases were separated by centrifugation at 3,000 rpm for 5 min. Only the organic phase (approximately 4 ml) was then transferred to a clean tube. The organic phase was then dried under the stream of nitrogen gas at 40°C. The residue was reconstituted by adding 200 ml of 0.1 M HCl and mixed by vortexing at full speed for 1 min. 80 ml of this acid solution was then analyzed by injecting on to a C18 column (LunaR, 150 x 4.6 mm i.d., 5 mm, Phenomenex, Torrance, CA, USA). The mobile phase was methanol: acetronitrile: disodium phosphate buffer (5 g/L) at ratio of 12: 24:64 v/v and adjusted pH to 4.0 by phosphoric acid. Flow rate of mobile phase was set at 1.3 ml/min and absorbance was monitored at 240 nm, AUFS 0.5. Standard curves were constructed in the clozapine concentrations range from 50-1000 ng/ml. The standard curve samples were treated in the same manner as the plasma samples collected from the volunteers. Clozapine concentrations in quality control and study samples were quantified by comparison of the peak height ratios between clozapine peak and internal standard peak with those of the standard curve.
Data analysis
Maximum observed plasma concentration (Cmax), Minimum observed plasma concentration (Cmin) and time of maximum plasma concentration (Tmax) were obtained from drug concentration-time curves. The area under the clozapine concentration-time curves from 0-12 h (AUC0-12h ) were calculated using the trapezoidal method and the first order elimination rate constant (Ke) was estimated using least square regression of points describing the terminal log-linear decaying phase. The terminal half-life (T1/2) was derived from ln2/Ke. The average concentration of drug in plasma during 12 h (Cav) was calculated from AUC0-12h (Cav = AUC0-12h/12). The percentage of fluctuation of drug level in plasma at steady state (%Fluctuation) was calculated using the following formula, % Fluctuation = 100 x (Cmax-Cmin)/Cav.
The differences in AUC0-12h, Cmax, Tmax and % Fluctuation between the reference product and the tested product were determined using ANOVA, two-way cross over design at confidence level of 95%. According to the standard criteria of the Health Canada and the United State Food and Drug Administration (US-FDA), bioequivalence of the two formulations was established when formulation or treatment effect of AUC0-12h and Cmax should not be different at alpha level of 0.05 and the 90% confidence interval of the mean ratio of AUC0-12h and Cmax between the tested product and the reference product should fell within the 0.80 to 1.25 for log-transformed data.
Results
The HPLC method used in this study is simple and provides appropriated sensitivity, specificity for the pharmacokinetic study of clozapine. Under described chromatographic conditions, the retention times for clozapine and internal standard were 5.8 and 12.7 min, respectively (Figure 1).
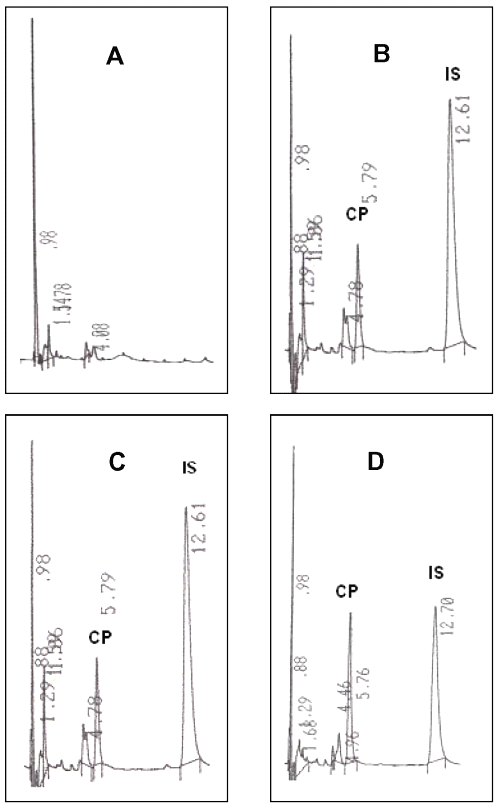
Figure 1: Typical chromatograms for determination of clozapine in plasma. A: Plasma blank; B: Plasma blank containing clozapine 200 ng/ml., C-D: Plasma obtained at day-7 after 30 min and 2 hr administration of 100 mg clozapine, respectively. CP: Clozapine peak, IS: Internal standard peak.
No endogenous interfering peak appeared at the retention times of the compounds of interest. Within-day coefficients of variation (determined in 5 replicated samples) were 8.92%, 1.21% and 4.74% at clozapine concentrations of 50, 100 and 400 ng/ml, respectively. Within-day accuracies were 108.09 ± 8.92%, 99.57 ± 1.21% and 104.56 ± 4.74% at 50, 100 and 400 ng/ml, respectively. While between-day coefficients of variation for clozapine (determined in 5 replicated samples) were 5.84%, 9.68% and 4.05% at 50, 100 and 400 ng/ml, respectively. Between-day accuracies were 107.80 ± 5.84%, 104.38 ± 9.68% and 99.95 ± 4.05% at 50, 100 and 400 ng/ml, respectively. The minimum quantifiable concentration for clozapine was set at 50 ng/ml. The calibration curves were linear over the range of 50-1000 ng/ml.
Determination the steady- state drug concentration
All of the participating patients were physically healthy based on both their medical examination and the results form clinical laboratory tests. From physical examination, laboratory tests and electrocardiogram revealed that none of the subjects suffered from any serious side effect from the clozapine products used in the study and all were well tolerated to both formulations. Hypersalivation and sedation were observed in some patients. The plasma clozapine concentrations in each subject at day 5-day-7 of each study phase prior to the morning dose of the drug were determined in order to confirm the steady-state conditions.
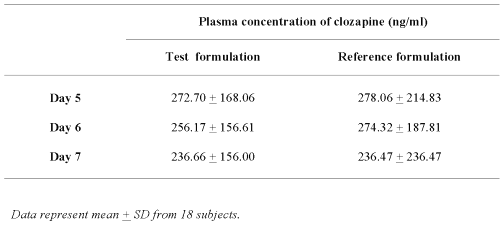
As shown in Table 1, the mean plasma clozapine levels at day-5, -6 and -7 prior the morning dose of either the Test or Reference formulation in 18 subjects were not significantly different.
Table 1: The mean plasma concentrations of clozapine in 18 subjects prior to administration of the morning dose.
Comparative pharmacokinetic parameters and bioequivalence evaluation
The mean clozapine concentration-time profiles after administration of the test and reference formulations in 18 subjects are depicted in Figure 2.
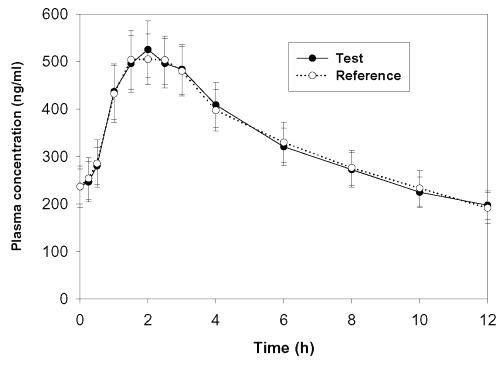
Figure 2: Mean plasma concentration-time profiles of clozapine in 18 subjects after oral administration of either a 100 mg Test or Reference formulation. Y- Bars represent SE values.
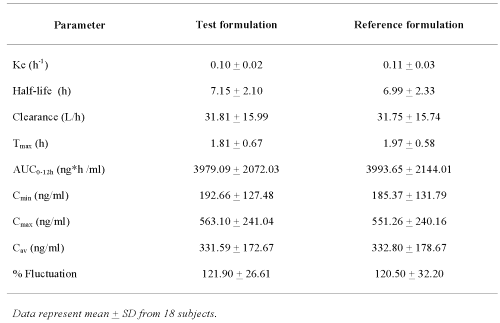
All of the pharmacokinetic parameters calculated for the Test formulation were close to those of the Reference formulation and there were no statistically significant differences between the two products (Table 2).
Table 2: Mean pharmacokinetic parameters of clozapine obtained from 18 subjects after administration of either Test or Reference formulation.
The ratio of the ln transformed data of both formulations was calculated for each subject (Table 3).
Table 3: Comparative mean ratio of ln- transformed Cmax and ln- transformed AUC0-12h of clozapine in 18 subjects after administration of Test formulation (T) and Reference formulation (R).
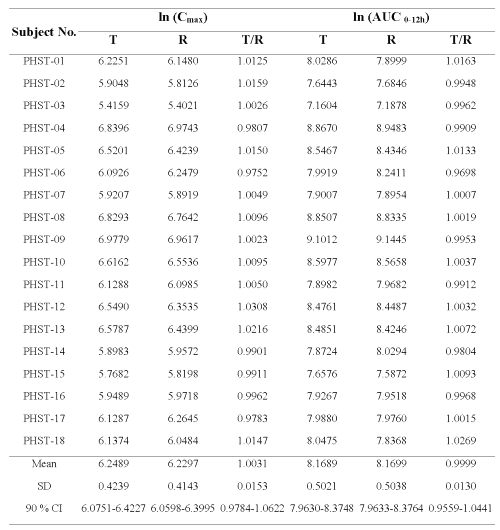
The mean of the ratio for lnCmax and lnAUC0-12h between the Test and Reference formulations were 1.0031+ 0.0153 and 0.9999 ± 0.0130, respectively. Analysis of variance (ANOVA) of the lnAUC0-12h and lnCmax obtained from the Test and Reference formulations revealed that the sequence, period or formulation was not significant different at p<0.05. However, inter-individual variation (subject factor) of these kinetic parameters was noticed. The 90 % confidence interval for lnAUC0-12h ranged from 0.9559 to 1.0441 and for lnCmax ranged from 0.9784 to 1.0622. It should also be noted that none of these values obtained from 18 subjects were outside the range of 0.8-1.25. In addition, ANOVA of lnTmax and % Fluctuation revealed that these two clozapine products were not significantly different from each other.
Discussions
Due to the hypotensive effects and serious bradycardia associated with the administration of clozapine to healthy subjects, the original recommendations of US-FDA for bioequivalence studies of generic clozapine products published in November 1996 has been revised. The most recent version of this guidance recommends that the bioequivalence study of this drug should be performed only on schizophrenic patients under multiple-dose steady-state condition (19). It has been reported that the pharmacokinetic parameters observed in the single and multiple-dose of clozapine were different. The mean terminal half-life values observed from the multiple-dose study were significantly higher than those observed in the single dose study (14.2 h vs. 7.9 h, respectively) (17). The elimination half-life increases significantly upon multiple dosing relative to single-dose administration, raising the possibility of concentration-dependent pharmacokinetics. However, linearly dose-proportional changes in AUC, peak, and minimum clozapine plasma concentrations after administration of 37.5 mg, 75 mg, and 150 mg twice daily have been observed (20). Although it is believed that the previously recommended study design using low dose (12.5 mg) in healthy subjects was adequate to establish bioequivalence of generic clozapine products, the recent guidance suggests the use of the highest dosage strengths for bioequivalence study of this drug in schizophrenic patients. Thus, the present study was carried out following the recent US-FDA Guidance using a multiple-dose, steady-state bioequivalence study in schizophrenic patients who received stable dose of 100 mg clozapine at 12 h interval.
Interindividual variability in pharmacokinetic parameters was observed among 18 male Thai Schizophrenic patients. The pharmacokinetic parameters such as Tmax, terminal half-life and total clearance (uncorrected for bioavailability) observed in the present study were comparable to those previously reports in schizophrenic patients under single-dose or multiple-dose, steady-state conditions (11, 21, 22).
Comparative pharmacokinetic study of the two clozapine products, Test formulation (Cloril) and Reference formulation (Clozaril) after multiple dosing regimen by administration of 100 mg bid in male schizophrenic patients revealed that the parameters which indicate the amount of drug absorbed into the body (AUC0-12h) and the relative rate of drug absorption (Cmax) obtained from these two formulations were very similar. The 90% confidence interval of the mean ratio of lnAUC0-12h was within 0.80-1.25 range which indicates that the Test formulation, Cloril was bioequivalent to the Reference formulation, Clozaril. Thus, it can be concluded that these two clozapine formulations when orally administered to schizophrenic patients were bioequivalent regarding rate and extent of absorption.
Several issues and concerns relevant to the switching of brand to generic clozapine have recently been raised. There are some controversies about an increase in the incidence of relapse of psychotic symptoms in stable Clozaril-treated patients after switching from brand-name clozapine to a generic clozapine (23-28). It should be noted that most of the studies were done under small size of the subjects, therefore large, controlled, prospective trials are needed to clarify the potential for treatment failure with the use of generic clozapine. In addition, evaluation of bioequivalence of generic clozapine under real-life conditions using a steady-state, multiple-dose bioequivalence study in schizophrenic patients may provide more direct therapeutic relevant to bioavailability finding.
Acknowledgements
We would like to thank the patients, nurses and all of the staff at Don Doo Rehabilitation Center, Khon Kaen Rajanagarindra Psychiatric Hospital, Khon Kaen for their collaboration. We thank Dr. Julraht Konsil, Faculty of Pharmaceutical Sciences for her skill in statistical analysis. This study was partially supported by Atlantic Laboratories Corp., Ltd., Bangkok, Thailand.
References
[1] Ereshefsky, L.; Watanabe, M.D. and Tran-Johnson, T., Clozapine: an atypical antipsychotic. Clin Pharm, 8: 691-709, 1989.
[2] Bladessarini, R.J. and Frankenburg, F.R., Clozapine: A novel antipsychotic agent. New Engl J Med, 324: 746-754, 1991.
[3] Meltzer, H.Y., Treatment-resistant schizophrenia-The role of clozapine. Curr Med Res Opin, 14: 1-20,1997.
[4] Kane, J.; Honigfeld, G.; Singer, J. and Meltzer, H., Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry, 45: 789-796, 1988.
[5] Fitton, A. and Heel, R.C., Clozapine: a review of pharmacological properties and therapeutic use in schizophrenia. Drugs, 5: 722-725, 1990.
[6] Richelson, E., Receptor pharmacology of neuroleptics: relation to clinical effects. J Clin Psychiatry, 60: 5-14, 1999.
[7] Lieberman, J.A., Understanding the mechanism of action of atypical antipsychotic drugs. A review of compounds in use and development. Br J Psychiatry, 22: 7-18, 1993.
[8] Jann, M.W.; Grimsley, S.R.; Gray, E.C. and Chang, W.H., Pharmacokinetics and pharmacodynamics of clozapine. Clin Pharmacokinet, 24: 161-176,1993.
[9] Ackenheil, M., Clozapine-pharmacokinetic investigations and biochemical effects in man. Psychopharmacol, 99: S32-37, 1989.
[10] Schaber, G.; Stevens, L.; Gaener, H.J.; Dietz, K. and Breyer-Pfaff, U., Pharmacokinetics of clozapine and its metabolites in psychiatric patients: plasma protein binding and renal clearance. Br J Clin Pharmacol, 46: 453-459, 1998.
[11] Cheng, Y.F.; Lundberg, T.; Bondesson, U.; Lindstrom, L. and Gabrielsson, J., Clinical pharmacokinetics of clozapine in chronic schizophrenic patients. Eur J Clin Pharmacol, 34: 445-449, 1988.
[12] Gauch, R. and Michaelis, W., The metabolism of 8-chloro-11-(4-methyl-1-piperazinyl)-5H-dibenzo-[b, e][1, 4]diazepine (clozapine) in mice, dogs and human subjects. Farmaco, 26: 667-681, 1971.
[13] Ereshefsky, L., Pharmacokinetics and drug interactions: update for new antipsychotics. J Clin Psychiatry, 57: 12-25, 1996.
[14] Pokorny, R.; Finkel, M.J. and Robinson, W.T., Normal volunteers should not be used for bioavailability or bioequivalence studies of cozapine. Pharm Res, 11: 1221, 1994.
[15] Levoyer, D.; Martinet, J.P.; Badiche, A. and Millet, B., Ten years of clinical experience with clozapine about 170 patients. Encephale, 30: 285-295, 2004.
[16] Hagg, S.; Spigset, O.; Bate, A. and Soderstrom, T.G., Myocarditis related to clozapine treatment. J Clin Psychopharmacol, 21: 382-388, 2001.
[17] Choc, M.G.; Hsuan, F.; Honigfeld, G.; Robinson, W.T.; Ereshefsky, L.; Crismon, M.L.;S aklad, S.R.; Hirschowitz, J. and Wagner, R., Single- vs. multiple-dose pharmacokinetics of clozapine in psychiatric patients. Pharm Res, 7: 347-351, 1990.
[18] Chung, M.C.; Lin, S.K. and Chang, W.H., Determination of clozapine and desmethylclozapine in human plasma by high performance liquid chromatography with ultraviolet detection. J Chromatogr, 613: 168-173, 1993.
[19] CDER, Center for Drug Evaluation and Research, US-FDA. Guidance for Industry: Clozapine tablets: In vivo bioequivalence and in vitro dissolution testing. 2003. http://www.fda.gov/cder/guidance/ index.htm (Accessed 10 Jan 2005)
[20] Choc, M.G.; Lehr, R.G.; Hsuan, F.; Honigfeld, G.; Smith, H.T.; Borison, R. and Volavka, J., Multiple-dose pharmacokinetics of clozapine in patients. Pharm Res, 4: 402-405, 1987.
[21] Lin, S.K.; Chang, W.H.; Chung, M.C.; Lam, Y.W. and Jann, M.W., Disposition of clozapine and desmethylclozapine in schizophrenic patients. J Clin Pharmacol, 34: 318-324, 1994.
[22] Guitton, C.; Abbar, M.; Kinowski, J.M.; Chabrand, P. and Bressolle, F., Multiple-dose pharmacokinetics of clozapine in patients with chronic schizophrenia. J Clin Psychopharmacol, 18: 470-476, 1998.
[23] Kluznik, J.C.; Walbek, N.H.; Farnsworth, M.G. and Melstrom, K., Clinical effects of a randomized switched of patients from clozaril to generic clozapine. J Clin Psychiatry, 62: 14-17, 2001.
[24] Mofsen, R. and Balter, J., Case reports of the reemergence of psychotic symptoms after conversion from brand -name clozapine to a generic formulation. Clin Ther, 23: 1720-1731, 2001.
[25] Sajbel, T.A.; Carter, G.W. and Wiley, R.B., Coverting patients from brand-name clozapine to generic clozapine. Ann Pharmacolther, 35: 281-284, 2001.
[26] Makela, E.H.; Cutlip, W.D.; Stevenson, J.M.; Weimer, J.M.; Abdallah, E.S.; Akhtar, R.S.; Aboraya, A.S. and Gunel, E., Branded versus geneic clozapine for treatment of schizophrenia. Ann Pharmacolther, 37: 350-353, 2003.
[27] Nuss, P.; Taylor, D.; De Hert, M. and Hummer, M., The generic alternative in schizophrenia: opportunity or treat? CNS Drugs, 18: 769-775, 2004.
[28] Layton, S. and Barbeau, M., Generic replacement of clozapine: a simple decision model from a Canadian perspective. Curr Med Res Opin, 20: 453-459, 2004.
Corresponding Author: Wichittra Tassaneeyakul, Department of Pharmacology, Faculty of Medicine, Khon Kaen University 40002, Thailand. wichitt@kku.ac.th
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.cspscanada.org