J Pharm Pharmaceut Sci (www.cspscanada.org) 8(1):18-25, 2005
The effect of type and concentration of vehicles on the dissolution rate of a poorly soluble drug (indomethacin) from liquisolid compacts.
Ali Nokhodchi1
Department of Pharmacy, School of Health and Life Sciences, King's College London, London, United KingdomYousef Javadzadeh, Mohammad Reza Siahi-Shadbad, Mohammad Barzegar-Jalali
Department of Pharmaceutics, School of Pharmacy, Tabriz University of Medical Sciences, Tabriz, IranReceived 10 November 2004, Revised 5 December 2004, Accepted 6 December 2004, Published 12 January 2005
PDF Version
Abstract
PURPOSE. For poorly soluble, highly permeable (Class II) drugs, such as indomethacin, the rate of oral absorption is often controlled by the dissolution rate in the gastrointestinal tract. Therefore together with the permeability, the solubility and dissolution behaviour of a drug are key determinants of its oral bioavailability. The object of the present study is to increase dissolution rate of indomethacin using liquisolid compacts. METHODS: Several formulations of liquisolid compacts containing various ratios of drug: propylene glycol (ranging from 1:1 to 1:4) was prepared. In this study the ratio of microcrystalline cellulose (carrier) to silica (coating powder material) was 20 in all formulations. The dissolution behaviour of indomethacin from liquisolid compacts and conventional formulations was investigated at different pHs (1.2 and 7.2). RESULTS: The results showed that liquisolid compacts demonstrated considerably higher drug dissolution rates than those of conventionally made capsules and directly compressed tablets containing indomethacin. This was due to increased wetting properties and surface of drug available for dissolution. Also it has been shown that the fraction of molecularly dispersed drug (FM) in the liquid medication of liquisolid systems was directly proportional to their indomethacin dissolution rates (DR). An attempt was made to correlate the percentage drug dissolved in 10-min with the solubility of indomethacin in different vehicles. A plot of the percentage drug dissolved against the solubility of indomethacin showed that the amount of drug dissolved increased linearly (correlation coefficient of 0.9994 and 0.996 at pH 7.2 and 1.2 respectively) with an increase in solubility of indomethacin in the vehicles. CONCLUSION: The liquisolid compacts technique can be a promising alternative for the formulation of water insoluble drugs, such as indomethacin into rapid release tablets.
Introduction
For poorly soluble, highly permeable (Class II) drugs, the rate of oral absorption is often controlled by the dissolution rate in the gastrointestinal tract (1). Therefore together with the permeability, the solubility and dissolution behaviour of a drug are key determinants of its oral bioavailability. There have been numerous efforts to improve drug dissolution rate. These include, (a) reducing particle size to increase surface area, thus increasing dissolution rate of drug; (b) solubilization in surfactant systems; (c) formation of water-soluble complexes; (d) use of pro-drug and drug derivatization such as a strong electrolyte salt forms that usually have higher dissolution rate; and (e) manipulation of solid state of drug substance to improve drug dissolution i.e. by decreasing crystallinity of drug substance through formation of solid solutions (2). The most common method is increasing the surface area of the drug by micronization. But, in practice the effect of micronization is often disappointing, especially when the drugs are encapsulated or tableted (3-5). This phenomenon was attributed to the agglomeration tendency of micronized, poorly soluble, hydrophobic drugs, which results in a decreased effective surface area for dissolution. The most promising method for promoting dissolution is the formation of liquisolid tablets (6-8).
The concept of liquisolid compacts, as defined by Spireas et al (6), can be used to formulate liquid medication such as oily liquid drugs and solutions or suspensions of water-insoluble solid drugs in non-volatile vehicles, into acceptably flowing and compressible powders. Using this new formulation technique, a liquid medication may be converted into a dry- looking, non-adherent, free flowing and readily compressible powder by a simple blending with selected powder excipients referred to as the carrier and coating materials. Various grades of cellulose, starch, lactose, etc, may be used as the carrier, whereas very fine particle size silica powder may be used as the coating material.
The antirheumatic agent, indomethacin, exhibits poor solubility (9). This undesirable physical property may increase the incidence of irritating side effects on the gastrointestinal tract because of a prolonged contact time with the mucosa (9). Numerous attempts (10-12) have been made to improve the dissolution rate of this widely used antirheumatic agent, to obtain more rapid and complete absorption. Thus it is an ideal candidate for testing the potential of rapid-release liquisolid compacts.
In this study indomethacin, a poorly water-soluble non- steroidal anti-inflammatory drug was formulated into 25 mg liquisolid compacts consisting of microcystalline cellulose, silica, and propylene glycol. The drug release was run using the No. 2 USP dissolution test apparatus at pHs 1.2 and 7.2 and the dissolution rates of liquisolid formulations were compared to those of conventionally prepared (hard gelatine capsule and directly compressed tablets). In this study the effect of type of vehicle on the dissolution rate of indomethacin from liquisolid compacts was also investigated.
Materials and Methods
Materials
Indomethacin was provided by Industrial Shahid Razakani (Tehran, Iran). Coarse granular microcrystalline cellulose (Mingtai Chemical, Taiwan), sodium starch glycolate (Yung Zip Chemical, Taiwan), nm-sized amorphous silicon dioxide (Mingtai Chemical, Taiwan), propylene glycol (Merck, Germany), PEG 400 (Merck, Germany) and Tween 80 (Merck, Germany) were used.
Preparation of conventional tablet, capsule, and liquisolid compacts
Indomethacin conventional tablets were produced by mixing the drug with microcrystalline cellulose-silica (ratio of microcrystalline cellulose: silica was 20:1) for a period of 10 min in a cubic mixer (Erweka, Type UG, Germany). The mixture was mixed with sodium starch glycolate for 10 min. The mixture was compressed on a 10-mm punch and die using a manual-tableting machine (Grasby, Riken, Japan) equipped with strain gauge (10-400 kg/cm2). Sufficient compression load between 80-100 kg/cm2 was applied in order to produce tablets hardness of 6-7 kp, as determined using a hardness tester (Erweka, TBH30MD, Germany). This formulation was denoted as DCT and each tablet contains 25 mg indomethacin, 100 mg of coarse granular microcrystalline cellulose, 5 mg of nm-sized silica and 5 mg sodium starch glycolate. Also a conventional capsule dosage form of micronized indomethacin prepared with the same composition, without disintegrating agent, as described above.
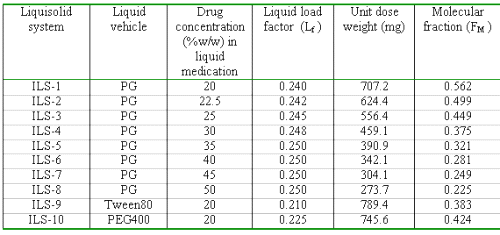
Several liquisolid of indomethacin compacts (denoted as ILS-1 to ILS-10) were prepared as follows. Indomethacin was dispersed in propylene glycol (this vehicle was used as the liquid vehicle to prepare the liquid medication of the different drug concentrations) with different ratios ranging from 1:1 to 1:4 (drug: propylene glycol). Then binary mixtures of microcrystalline cellulose-silica (microcrystalline cellulose as the carrier powder and silica as the coating material with a ratio of 20, R) were added to the mixture containing the drug and propylene glycol under continuous mixing in a mortar. Depending on the ratio of drug: propylene glycol in the liquid medication used, different liquid load factors (the liquid load factor, Lf, is the weight ratio of the liquid medication and carrier powder in the liquid solid formulations) were employed in our liquisolid preparations. These amounts of the carrier and coating materials are enough to maintain acceptable flow and compression properties. Sodium starch glycolate (5% w/w) as a disintegrant was mixed with all formulations for a period of 10 min. The final mixture was compressed using the manual tableting machine to achieve tablet hardness of 6-7 kp. In order to investigate the effect of vehicle type, Tween 80 and PEG 400 were used at a ratio of 1:4 (indomethacin: vehicle). Important formulation characteristics of the prepared indomethacin liquisolid compacts are shown in Table 1.
Table 1: Key formulation characteristics of prepared indomethacin liquisolid compacts.
Spectrophotometeric Analysis
The spectrophotometeric analysis of all indomethacin samples in aqueous solutions (pH 1.2 or 7.2) were performed at 318 nm (UV/Visible spectrophotometer, Shimadzu-120, Japan). Standard curves were constructed by serially diluting an aqueous stock solution of the drug (at pHs 1.2 and 7.2) to obtain concentrations in the range of 3.125-50 mg/ml using simulated gastric fluid (SGF) or simulated intestine fluid (SIF) as the diluents. Each sample was analysed in triplicate.
Solubility studies
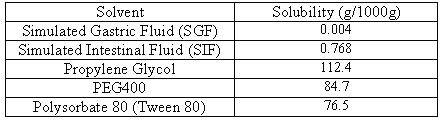
Solubility measurements were performed according to the method of Higuchi and Connors. In brief, solubility studies of indomethacin were carried out in SGF, SIF, propylene glycol, PEG 400 and Tween 80. Saturated solutions were prepared by adding excess drug to the vehicles and shaking on the shaker (Velp, Italy) for 48h at 25 ± 0.5°C under constant vibration. After this period the solutions were filtered, diluted and analysed by UV-spectrophotometer (Shimadzu, Japan). Three determinations were carried out for each sample to calculate the solubility of indomethacin.
Dissolution studies
The USP paddle method (Erweka, DPT6R, Germany) was used for all the in vitro dissolution studies. In this method, distilled water which simulated gastric fluid (pH 1.2), and intestinal fluid (pH 7.2) without enzyme, were used as dissolution media. The rate of stirring was 100 ± 2 rpm. The amount of indomethacin was 25 mg in all formulations. The dosage forms were placed in 900 ml of gastric fluid or intestinal fluid and maintained at 37 ± 0.1°C. At appropriate intervals (10, 20, 30 and 60 min), 5 ml of the samples were taken and filtered through a 0.45 mm Millipore filter. The dissolution media was then replaced by 5 ml of fresh dissolution fluid to maintain a constant volume. The samples were then analysed at 318 by UV/visible spectrophotometer. The mean of at least three determinations was used to calculate the drug release from each of the formulations.
In each dissolution test, a standard solution of indomethacin (in the medium used) that had a concentration equal to the one expected to be obtained at the 100% dissolution level, was placed in the seventh dissolution vessel of set. In each sample, the percent of dissolved drug was obtained by comparing its spectrophotometric absorbance (Absample) with absorbance of the standard solution (Abstandard) as follows:
Drug dissolved (%) = (Absample / Abstandard) c 100
For assessment and comparison, drug dissolution rates (DR) were used. To this end, the amount of drug (in mg) dissolved per minute during the first 10 min, was calculated as follows;
DR = (M ∞ D)/1000
Where M is the total amount of indomethacin in each tablet (in this study it is 25000 mg) and D denotes percentage of drug dissolved during the first 10 min.
Statistical analysis
All the data were statistically analysed by analysis of variance or Tukey's multiple comparison test. A linear regression analysis was used to test associations between two parameters. Results are quoted as significant where p < 0.05.
Results
The results of solubility measurements are presented in Table 2.
Table 2: Solubility of indomethacin in various solvents.
The solubility of indomethacin (pKa = 4.5) in pH 1.2 and pH 7.2 buffered aqueous medium at 25°C was found to be 3.882 mg/ml and 767.5 mg/ml, respectively. Therefore, according to the USP solubility definition, indomethacin can be considered as a practically insoluble drug at pH 1.2 and slightly soluble at pH 7.2. The results in Table 2 show that the solubility of indomethacin is considerably increased in presence of propylene glycol, PEG 400 or Tween 80. For instance, the increase in solubility of indomethacin at 25°C in propylene glycol was about 28969- and 145-fold compared to pure indomethacin at pH 1.2 and 7.2, respectively. Figure 1 shows the dissolution profiles of indomethacin from the liquisolid compacts (ILS-1, ratio of drug: poropylene glycol is 1:4) and conventional formulations (tablets and capsules) at different pHs.
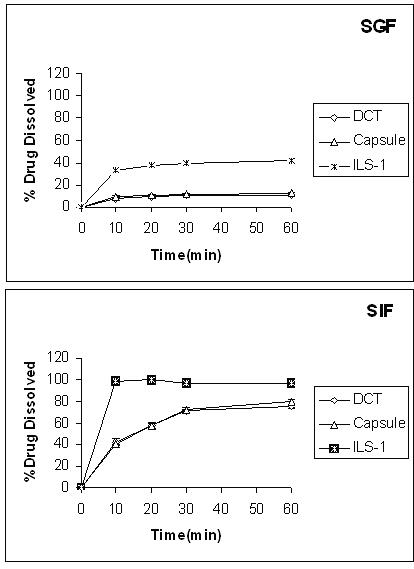
Figue 1: Dissolution profiles of indomethacin liquisolid compacts (ILS-1), conventional capsule and directly compressed tablets (DCT) at different dissolution media. Error bars are standard deviations for at least 3 determinations.
This Figure shows that liquisolid compacts produce higher dissolution rate in comparison with conventional dosage forms. Figure 2 shows the DR of indomethacin from ILS-1, ILS-8 (the highest and lowest concentrations of propylene glycol in formulations respectively), conventional capsule and directly compressed tablet in first 10 min.
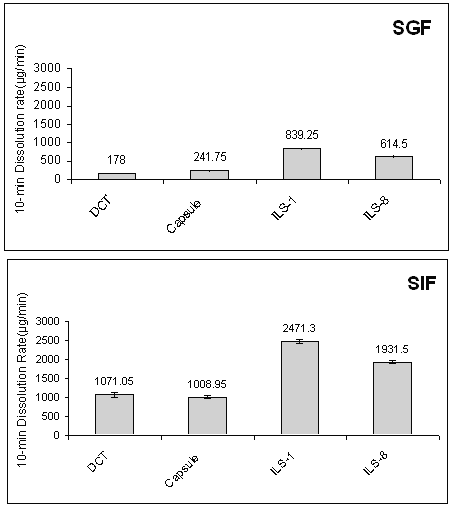
Figure 2: Comparison of the 10-min dissolution rate of indomethacin exhibited by liquisolid compacts (ILS-1), conventional capsule and directly compressed tablets (DCT) at different dissolution media. Error bars are standard deviations for at least 3 determinations.
As it is clear from this figure, at any dissolution media the liquisolid tablets displayed better in-vitro release characteristics than those of the directly compressed and capsule counterparts (p < 0.05). Figure 3 shows the effect of drug concentration (Cd) in the liquid medication on the 10 min dissolution rate (DR) of indomethacin from the propylene glycol liquisolid compacts in SGF and SIF media.
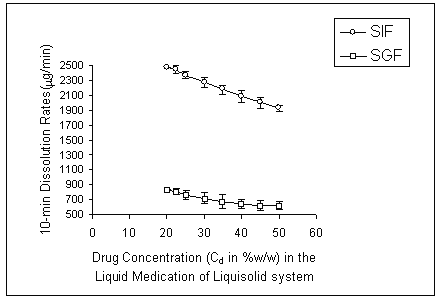
Figure 3: Effect of drug concentration (Cd) in the liquid medication on the 10-min dissolution rate (DR) exhibited by different liquisolid formulations at various dissolution media (error bars are standard deviations for at least 3 determinations).
It can be seen that, as the concentration of drug in liquid medication (Cd) increased from 20 to 50% w/w, the values of DR decreased.
In order to investigate the effect of fraction of the dissolved or molecularly dispersed of indomethacin (FM) in the liquid medication of the prepared liquisolid tablets on the dissolution rates of the drug from liquisolid compacts, conventional capsules and DC tablets, the FM was plotted against their corresponding DR values (Fig. 4).
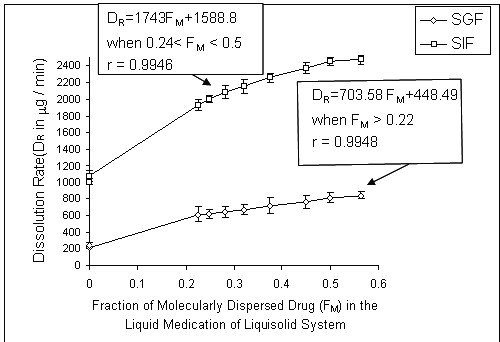
Figure 4: Effect of the fraction of molecularly dispersed drug (FM) in the Liquisolid systems on the 10-min dissolution rate (DR) of indomethacin exhibited by various liquisolid formulations at different dissolution media. Error bars are standard deviations for at least 3 determinations.
FM can be defined as the ratio of the drug's saturation solubility (CL) in the liquid vehicle over the drug concentration (Cd) in the liquid medication. Therefore, FM = CL / Cd , where FM =1 and when CL / Cd > 1. Based on the above equation, the FM values of our formulations are listed in Table 1. Since no liquid vehicle is involved in the case of capsules and directly compressed tablets which contain plain indomethacin powder, their FM value was taken equal to 0.
The effect of the type of vehicle on the release rate of indomethacin from liquisolid compacts is shown in Fig. 5. It can be seen from the Figure, all 3 liquid medications (prepared with propylene glycol, PEG 400 and Tween 80) used in liquisolid compacts produced higher dissolution rate than those of conventional tablet (DCT) or capsule formulations.
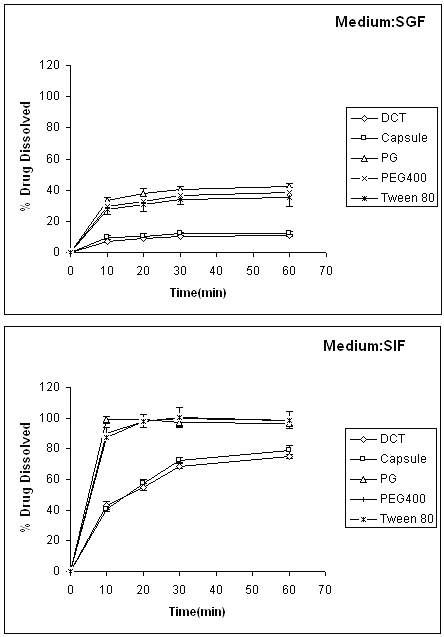
Figure 5: Dissolution profiles of indomethacin from liquisolid compacts prepared with different vehicles at different dissolution media. Error bars are standard deviations for at least 3 determinations.
Discussion
In vitro dissolution profiles of indomethacin showed that there was no significant difference (p > 0.05) between release rate of indomethacin from conventional tablet and capsule formulations at different pHs. Comparing the release profiles of indomethacin from liquisolid compacts and conventional dosage forms showed that the liquisolid formulations produced the highest dissolution rate at pH 1.2 and 7.2. Figure 1 also showed that the amount of drug release at pH 7.2 (SIF) is greater than those of released at pH 1.2 (SGF). This is due to the higher solubility of indomethacin at higher pH value (Table 2). The reason for an increase in dissolution rate of indomethcin from liquisolid compact can be explained by solubility data (Table 2). The solubility of indomethacin in SGF, SIF, propylene glycol, PEG 400 and Tween 80 is given in Table 2.
This table shows that the solubility of indomethacin can remarkably be improved in presence of propylene glycol.
It has been shown that at any dissolution media the liquisolid compacts displayed higher DR values. This indicates that the drug concentration in the liquid medication is one of the main influences on the performance of a liquisolid compact and has considerable effect on the indomethacin 10 min dissolution rate. Such differences in the DR values of indomethacin from liquisolid compacts observed in Fig. 3 may be justified using the solubilization and molecular dispersion states of the drug in the formulations. For instance, since the saturation solubility of indomethacin in propylene glycol is 11.24% w/w (Table 2), about 56.20% of the drug is in soluble form in ILS-1 formulation. Whereas in ILS-8 formulation about 22.4% of indomethacin is in soluble form in propylene glycol liquisolid compacts (ILS-8 formulation containing 50% indomethacin and 50% propylene glycol, whereas ILS-1 containing 20% indomethacin and 80% propylene glycol).
Such higher drug dissolution rates displayed by liquisolid compacts may also imply enhanced oral bioavailability. For instance, Khaled et al showed that the absolute bioavailability of hydroclorotiazide from the liquisolid tablets was 15% higher than that of the commercial tablets (13).
It can be seen from Fig. 4, in SGF or SIF media, there is a direct relationship between FM and DR values. The regression analyses showed that the DR in 10-min increased in a linear manner with increasing FM values of the liquisolid systems at different pHs. Therefore, it is possible to predict the dissolution rate (D R in mg/min) of indomethacin in propylene glycol liquisolid tablets, which will be obtained within the initial 10 min of the dissolution process, conducted using 100 rpm as the paddle speed and 900 ml of SGF or SIF as the dissolution media. A plot of the DR against FM (when 0.24 < FM < 0.50 and FM >0.22 at SIF and SGF respectively) shows that the DR changes linearly with FM . The high correlation coefficients of 0.9946 and 0.9948 provide a value that characterizes the effect of FM on indomethacin dissolution from propylene glycol liquisolid compacts at SGF and SIF respectively.
As shown in Fig. 4, for FM values greater than 0.22 and ranging from 0.24 to 0.50 at pH 1.2 and 7.2, respectively, the 10-min dissolution rate of indomethacin may be predicted by the following equations.
DR = 703.58 FM + 448.49 in SGF medium (pH=1.2)
DR = 1743 FM + 1588.8 in SIF medium (pH=7.2)
According to the equation (14), DR = (D/h) S (Cs-C), the drug dissolution rate (DR) is directly proportional to its concentration gradient (Cs-C) in the stagnant diffusion layer and its surface (S) available for dissolution. Cs is the saturation solubility of the drug in the dissolution medium and thus, it is a constant characteristic property related to the drug and dissolving liquid involved. Since all of dissolution tests for formulations were done at a constant rotational paddle speed (100 rpm) and identical dissolving media, we can assume that the thickness (h) of the stagnant diffusion layer and the diffusion coefficient (D) of the drug molecules remain almost identical. Since part of the drug is dissolved in co-solvents within liquisolid compacts. After tablet disintegration, the liquisolid primary particles suspended in the dissolving medium contain the drug in a state of molecular dispersion, whereas the directly compressed tablets or conventional capsule formulation are merely exposing micronised drug particles. In other words, in the case of liquisolid compacts, the surface of drug available for dissolution is related to its specific molecular surface which, by all means, is much greater than that of the indomethacin particles delivered by the plain, directly compressed tablets or the conventional capsule formulations. Therefore, the observed higher dissolution rates of indomethacin from liquisolid tablets are due to the significant increase in the specific surface area of indomethacin.
In addition, the saturation solubility of the drug in the microenvironment (Cs) might be increased in the liquisolid systems. In fact, the small amount of liquid vehicle contained per liquisolid compact, are not sufficient to increase the overall saturation solubility of drug in the aqueous dissolution medium. However, the solid/liquid interface between an individual liquisolid primary particle and the dissolving fluid involves particle surface to form the stagnant diffusion layer. In this microenvironment, it is possible that the infinite amounts of liquid vehicle that diffuse with the drug molecules out of a single liquisolid particle might be adequate to enhance the solubility of drug that act as a cosolvent with the dissolution medium of the stagnant diffusion layer. In result, such an increase in Cs, in a larger drug concentration gradient, increase the dissolution rate (7, 8, 15).
Figure 5 shows that liquisolid compacts containing propylene glycol produce higher dissolution rate in comparison with other liquisolid compacts containing PEG 400 or Tween 80 with the same concentration. For example, the percentages amount of the drug released from liquisolid compacts in 10 min in presence of propylene glycol, PEG 400 or Tween 80 were 34, 28 and 27 or 99, 90 and 87% at pH 1.2 or 7.2 respectively. This is due to a difference in solubility of indomethacin in these liquid medications (see Table 2).
An attempt was made to correlate the percentage drug released in 10 min with the solubility of indomethacin in different vehicles. A plot of the percentage drug released against the solubility of indomethacin shows that the amount of drug released changes linearly (correlation coefficient of 0.9994 and 0.996 at pH 7.2 and 1.2 respectively) with the solubility of indomethacin (Fig. 6).
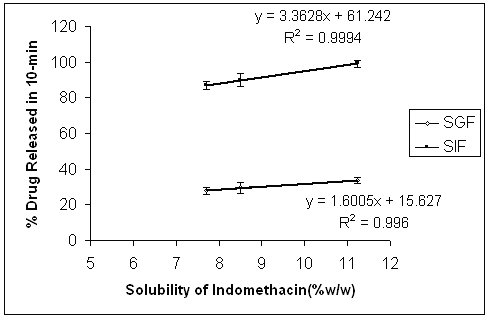
Figure 6: Regression lines showing the linear relationship between drugs released (%) and the solubility of indomethacin. Error bars are standard deviations for at least 3 determinations.
Regression analysis showed that the relationships between percentage release of the drug and the solubility in both media (SIF and SGF) was significant (p < 0.05 in ANOVA). Although the line for SGF medium has a much smaller slope (Fig. 6), it is still statistically significant (p < 0.05 in ANOVA). However it should be noted that the difference between release rates of the second and the last point at SGF medium was not statistically significant in Tukey's test (p > 0.05). These findings could be of value when deciding on the type of liquid medication to be used in liquisolid compact formulations
Conclusion
The liquisolid compacts technique can be a promising alternative for the formulation of water insoluble drugs, such as indomethacin into rapid release tablets. The higher dissolution rates displayed by liquisolid compacts may also imply enhanced oral bioavailability due to the increased wetting properties and solubility of drug in the liquid vehicles. It has been shown that the solubility of the drug in the liquid medication of the liquisolid compacts is directly proportional to their indometacin dissolution rates.
References
Lobenberg R, Amidon GL Modern bioavailability, bioequivalence and biopharmaceutics classification system; new scientific approaches to international regulatory standards. Eur. J. Pharm. Biopharm., 50: 3–12, 2000.
Kapsi SG, Ayres JW. Processing factors in development of solid solution formulation of itraconazole for enhancement of drug dissolution and bioavailability. Int. J. Pharm.; 229: 193–203, 2001 .
Aguiar AJ, Zelmer AJ, Kinkel AW. Deagglomeration behavior of relatively insoluble benzoic acid and its sodium salt. J. Pharm. Sci.; 56:1243–1252, 1967.
Finholt P, Solvang S. Dissolution kinetics of drugs in human gastric juice the role of surface tension. J. Pharm. Sci.; 57:1322–1326, 1968.
Lin SL, Menig J, Lachman L. Interdependence of physiological surfactant and drug particle size on the dissolution behavior of water insoluble drugs. J. Pharm. Sci.; 57: 2143–2146, 1968.
Spireas S, Sadu S. Enhancement of Prednisolone dissolution properties using liquisolid compacts. Int.. J. Pharma.; 166: 177-188, 1998.
Spireas S, Sadu S, Grover R. In vitro release evaluation of hydrocortisone liquisolid tablets, J. Pharm. Sci.; 87: 867-872, 1998.
Spireas S, Wang T, Grover R. Effect of powder substrate on the dissolution properties of methchrothiazide liquisolid compacts. Drug Dev. Ind. Pharm.; 25: 163-168, 1999.
Alsaidan SM, Alsughayer AA, Eshra AG. Improved dissolution rate of indomethacin by adsorbents. Drug Dev. Ind. Pharm.; 24 (4): 389–394, 1998.
Krasowska H. Solubilization of indomethacin and cinmetacin by non-ionic surfactants of the polyoxyethylene type. Il Farmaco; 31 (9): 463–472, 1976.
Krasowska, H. Effect of micellar solubilization on the gastrointestinal absorption of indomethacin in the rat. Int. J. Pharm.; 7: 137–143, 1980.
Habib MJ, Akogyeram C, Ahmadi B. Improved dissolution of indomethacin in coprecipitates with phospholipids. Part 1. Drug Dev. Ind. Pharm.; 19 (4): 499, 1993.
Khaled K.A., Asiri Y.A., El-Sayed Y.M. In vivo evaluation of hydrochlorothiazide liquisolid tablets in beagle dogs. Int. J. Pharm.; 222: 1-6, 2001.
Noyes AA, Whitney WR. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc.; 19: 930-934, 1897.
El-Moneim Darvish I, El-Kamal A. H. Dissolution enhancement of glibenclamide using liquisolid tablets technology. Acta Pharm. 51:173-181, 2001.
Corresponding Author: Ali Nokhodchi, Department of Pharmacy, School of Health and Life Sciences, King’s College London, 150 Stamford Street, Franklin-Wilkins Building, London SE1 9NN, United Kingdom. ali.nokhodchi@kcl.ac.uk
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.cspscanada.org