J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 7(3):378-388, 2004
Effect of acidic ternary compounds on the formation of miconazole/cyclodextrin inclusion complexes by means of supercritical carbon dioxide.
Valéry Barillaro1, Pascal Bertholet, Sandrine Henry de Hassonville
Laboratory of Pharmaceutical Technology, Department of Pharmacy, University of Liège, CHU-Tour 4, Bat. B36, Avenue de l'Hôpital, 1, 4000 Liège 1, BelgiumEric Ziemons
Laboratory of Analytical Chemistry, Department of Pharmacy, University of Liège, CHU-Tour 4, Bat. B36, Avenue de l'Hôpital, 1, 4000 Liège 1, BelgiumBrigitte Evrard, Luc Delattre, Géraldine Piel
Laboratory of Pharmaceutical Technology, Department of Pharmacy, University of Liège, CHU-Tour 4, Bat. B36, Avenue de l'Hôpital, 1, 4000 Liège 1, BelgiumReceived 27 August 2004, Revised 3 November 2004, Accepted 15 November 2004, Published 30 November 2004
PDF Version
Abstract
PURPOSE: The aim of the study is to evaluate the effect of different acidic compounds on the inclusion of miconazole (MICO) in several cyclodextrins (CDs) using supercritical carbon dioxide (SCCO2 ) processing. METHODS: Physical mixtures were processed by SCCO2 at 30 MPa, 125°C during 60 minutes in a static mode to produce inclusion complexes. The inclusion complexes were characterized by differential solubility, Fourier transform infrared spectroscopy (FT-IR) and dissolution test. RESULTS: The best inclusion yields were achieved with the combination of MICO base and HPγCD with or without acids. Maleic and fumaric acids influenced the MICO inclusion differently in function of their conformation. During the process, a miconazole salt was observed with maleic acid and characterized by thermal analysis and mass spectrometry. The kinetics inclusion followed a saturation-type shape curve. FT-IR confirmed the presence of genuine inclusion complexes. The complexes MICO base/HPγCD/(L-tartaric acid) enhanced the dissolution rates of MICO more than the corresponding physical mixtures did. Lastly, the stability study revealed that the complexes were stable. CONCLUSIONS: The formation of stable complexes between MICO and CDs is possible using SCCO2. Moreover an acidic ternary compound is able to modify the formation of the complex. The inclusion complexes, which show better dissolution profiles than those with the corresponding physical mixtures, could lead to an increase of the oral bioavailability of MICO.
Introduction
CDs have been used in pharmaceutical technology for many years as solubilizing agents1. Although CDs can increase the aqueous solubility of drugs, many applications need large amounts of CDs. But for several reasons, such as production costs and toxicity, CD amounts have to be reduced in pharmaceutical use. To achieve this goal, several approaches can be considered. The first is the use of chemically modified CDs, which present a higher solubility in water. The second method consists in adding a water-soluble polymer, like PVP or methylcellulose, with the aim to increase the solubility of both the complex and the drug itself2. The third method is the use of an acidic ternary compound. In the case of a basic drug, the acidic agent, for instance, a hydroxyacid, promotes the solubilization of the guest molecule both by forming a salt and by increasing the stability constant of the complex3. The same approach could be applied to an acidic drug by adding a basic ternary compound4.
SCCO2 has been successfully used for the preparation of inclusion complexes with CDs. This innovating process allows the production of solid inclusion complexes without the use of water or organic solvents. Van Hees et al.5 were able to prepare inclusion complexes with piroxicam, an anti-inflammatory non steroidal drug, and bCD by processing a physical mixture with SCCO2 (150°C, 45 MPa during 180 minutes). The presence of an inclusion compound was proved by DSC and FT-IR. More recently, Charoenchaitrakool et al. produced inclusion complexes between ibuprofen and methyl- bCD6. The ibuprofen content in the product corresponded to a 1:1 complex, and DSC found no crystalline ibuprofen.
MICO (1-2-((2, 4-dichlorophenyl)-2-(2, 4-dichlorophenyl)-methoxy) ethyl)-1-imidazole) is an antifungal drug which presents poor water solubility. Its pharmacological indications are limited to the treatment of intestinal and dermatological mycosis. Previously, MICO was also administered intravenously for the treatment of systemic fungal infections in the form of a micellar solution, with the drawback that this solution contains a surfactant, polyoxyl-35 castor oil, able to induce anaphylactoid reactions. Several CDs ( b CD, HP b CD, SBE-7-b CD, γCD, HPγCD...) have been used to complex MICO into their cavity7-9. The influence of a ternary acidic compound in solution was studied and the results showed a synergic effect between the CDs and the acid on the solubilization of MICO; this effect is independent of the pH value. This property allowed the development of an intravenous non-surfactant solution10.
In this study, the effect of both the CD type and the acidic ternary agent on the formation of solid inclusion complexes with MICO by means of SCCO2 will be investigated. In a second step, the influence of the conformation of the ternary compound will be determined. In a third step, the effect of process time on the inclusion will be studied. Then, the formation of inclusion complexes will be confirmed by FT-IR spectroscopy, differential solubility and dissolution studies.
Experimental Section
Materials
MICO (m.p. 84.3°C, Eur. Ph. 4th Edition) was obtained from Janssen Pharmaceutica (Beerse, Belgium). MICO nitrate (m.p. 184.1°C, Eur. Ph. 4th Edition) was purchased from Bufa (Uitgeest, Holland), bCD (Eur. Ph. 4th Edition, 7.58% H2O) and HPb CD (3.22 % H2O, D.S. 0.63) were kindly supplied by Roquette (Lestrem, France). γ CD (4.25 % H2O) and HP γ CD (1.62 % H2O, D.S. 0.58) were obtained from Wacker Chemie GmbH (Munich, Germany). Citric acid (m.p. 153°C, pKa1 3.13, pKa2 4.76, pKa3 6.40, Eur Ph. 4th Edition) and L-tartaric acid (m.p. 206°C, pKa1 2.93, pKa2 4.23, Eur Ph. 4th Edition) were purchased from Merck (Darmstadt, Germany), malic acid (m.p. 131°C, pKa1 3.46, pKa2 5.10) from Sigma-Aldrich (Steinhem, Germany), fumaric acid (m.p. 287 °C, pKa1 3.10, pKa2 7.80) from Fluka (Buchs, Switzerland) and maleic acid (m.p. 139°C, pKa1 1.90, pKa2 6.70) from Acros (New Jersey, USA). The chemical structure of all these compounds is depicted in figure 1.
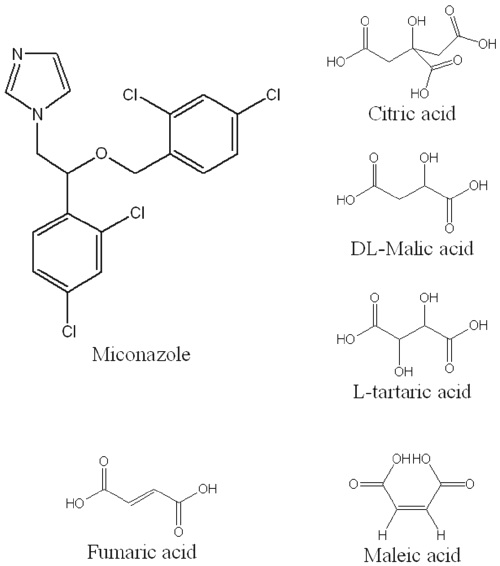
Figure 1: Chemical structures of MICO and the used acids.
R14821 (2, 4-dichlorophenyl)-1H-imidazol-1-ethanol) were purchased from Sigma-Aldrich (Steihnhem, Germany) and R66716 (2, 4-dichlorobenzenemethanol) from Acros (New Jersey, USA). CO2 was of N48 quality from Air Liquide (99.998%) (Liège, Belgium). All other products were of analytical grade.
Methods
Preparation of physical complexes
The physical mixtures were prepared by grinding, in a mortar, calculated and exactly weighed amounts of MICO, CDs and acid in equimolar ratio.
Preparation of inclusion complexes
The inclusion experiments with SCCO2 were performed with a SUPREX SF Extractor Autoprep 44 (Pittsburgh, PA, USA), described in previous works5. A 1-ml vessel was filled with 400 or 600 mg of physical mixtures of MICO-CD 1:1 (mol:mol) or MICO-CD-acid 1:1:1 (mol:mol:mol). The content was pressurized (± 1 minute) and left in a static mode during 60 minutes at 30 MPa and 125°C. At the end of the experiment, the vessel was depressurized within 15 seconds. The vessel content, in the form of a compact solid, was emptied, ground and homogenized in a mortar before further analysis. The kinetics of the formation of the inclusion complexes were also studied by processing physical mixtures with SCCO2 as described above during various contact times (15, 30, 60, 120 and 240 minutes). The effect of an acidic ternary compound was also determined.
Assay of miconazole
MICO was assayed by an HPLC method. The HPLC system consisted of an L-6000 Merck-Hitachi pressure pump, an L-7200 Merck-Hitachi autosampler, an L-7350 Merck-Hitachi column oven, an L-7400 Merck-Hitachi UV detector and a D-7000 interface. The system was controlled by a computer running the "HPLC System Manager v 4.0" acquisition software developed by Merck-Hitachi. Twenty-ml samples were injected into a Lichrocart column (125 x 4 mm i.d.) prepared with an octylsilane (C8) phase Lichrospher 60 RP-Select B 5 m m (Merck) and maintained at 30°C. The mobile phase consisted of a 70:30 (v/v) mixture of methanol (HPLC grade) and a 50 mM pH 3.5 ammonium acetate buffer. The flow rate was adjusted to 1.0 ml/min. All samples were analyzed twice. The detector was set at 230 nm. This method was validated11 and showed good linearity, reproducibility and accuracy between 5 and 80 m g/ml. Limits of detection (LOD) and quantification (LOQ) were determined and found to be 0.4 m g/ml for the LOD and 1.33 m g/ml for the LOQ.
Determination of inclusion yields
The inclusion yields were determined by a differential solubility method according to Van Hees et al.12. This method includes two steps. The first consists in the solubilization of the physical mixture or the complex in a solvent that dissolves the MICO, the CD and the ternary agent. This step allows determining total MICO content. The second step consists in the solubilization of the free MICO with an organic solvent in which the CD is insoluble and in which the included drug remains entrapped. So the total MICO content was determined by dissolving an exactly known quantity of MICO complex or physical mixture in the HPLC mobile phase. The MICO contained in the resulting solution was assayed using the HPLC method described above. In the same way, the free MICO content was measured by dispersing an exactly known quantity of MICO complex or physical mixture in acetonitrile. The suspension was sonicated (Transsonic T460) for five minutes, filtered through a Millex-GV 0.22 m m filter (Micropore) and analyzed by HPLC after appropriate dilution in order to determine the free MICO concentration. Determination of the total MICO content in the complexes produced by supercritical processing allows assessing that there is no loss of MICO during this process. In order to validate the differential solubility method, physical mixtures between MICO base and b CD, and between MICO base and γ CD, were prepared by gently grinding the powders in a mortar with a pestle. For each combination, the MICO base concentrations were 1.79 % (w/w) and 28.57 % (w/w) to reach final concentrations of 5 and 80 m g/ml of MICO base respectively. Two reference inclusion complexes were prepared. The first was a MICO base/b CD inclusion complex obtained by a precipitation method described by Pedersen et al.8. 16 mg/ml b CD were dissolved and 0.4 mg/ml MICO base were dispersed in a 50 mM pH 10 phosphate buffer. The mixture was stirred during 15 days at 4°C. After this time, the suspension was filtered and the insoluble fraction was kept in an oven at 60°C until dryness. The second complex was a MICO base/γ CD inclusion complex prepared according to Jacobsen et al.9. 100 mg/ml γ CD were dissolved and 1 mg/ml MICO base was dispersed in a 50 mM pH 7 phosphate buffer. The suspension was stirred at room temperature during 15 days. After this time, the suspension was filtered and the insoluble fraction was kept in an oven at 60°C until dryness. The complete MICO inclusion in both complexes was checked by DSC.
Infrared spectroscopy
The spectra of MICO, CDs, acids, physical mixtures and complexes were obtained from a KBr disk with a Perkin-Elmer GX FT-IR spectrophotometer (Beaconsfield, United Kingdom). The frequency range lies between 4000 and 450 cm-1.
Differential scanning calorimetry (DSC)
DSC experiments were carried out with a Mettler-Toledo DSC25-TC15 TA controller supervised by a computer running the "Star System v.6.10 SW" software. The experimental conditions were as follows: temperature range from 30 to 250°C with a heating rate of 10°C/min, pierced aluminum crucibles with about 6 mg product, nitrogen atmosphere with a 20 ml/min flow rate.
Liquid chromatography-Mass spectrometry (LC-MS) conditions
The HPLC is an HP 1000 series consisting of a binary pump, a vacuum degasser, a thermostated column compartment and an autosampler, all from Agilent Technologies (Waldbronn, Germany). The HPLC separations were performed at 30°C on a Waters RP-C8 Symmetry column (5 mm, 125 x 4 mm i.d.) (Milford, Massachusetts, U.S.A.). The mobile phase was a mixture of methanol and of a 50 mM pH 3.5 ammonium acetate buffer (70:30 v/v). The flow rate was 1 ml/min. and the injected sample volume was 20 ml. Mass spectrometry detection was carried out using a Micromass Ultima Quadrupole instrument (Manchester, United Kingdom) configured with a Z-spray electrospray ionization source and controlled by a computer running the "Masslynx v. 3.5" acquisition software. Source conditions were as follows: positive ion electrospray; capillary voltage: 3 kV; cone voltage: 14 V; source temperature: 145°C; desolvatation temperature: 450°C; cone gas flow (nitrogen): 94 l/h; and desolvatation gas flow (nitrogen): 552 l/h. Sample solutions were prepared by dissolving the compound in the mobile phase to reach the concentration of 10 mg/ml.
Dissolution studies
The dissolution profiles of MICO, physical mixtures and complexes were performed according to the rotating paddle method described in the 4th Edition of the European Pharmacopoeia. A dissolution test apparatus, Sotax AT7 (Basel, Switzerland), connected via a Waston-Marlow 505Du peristaltic pump (Falmouth, United Kingdom) to a Hitachi 3000 flow-through UV spectrophotometer (Tokyo, Japan) was used in this method. A quantity equivalent to 13 mg of MICO base was exactly weighed, introduced into a hard gelatin capsule, and then immersed by means of a sinker into 900 ml of dissolution medium containing a 50 mM pH 6.8 phosphate buffer. This medium was stirred at 100 rpm and set at 37 ± 0.1°C. The medium was pumped at a rate of 15 ml/min every 2 minutes, passed through a 0.47-mm filter and the absorbance of this solution was measured at 230 nm. Each experiment was carried out in duplicate. The standard solution at the concentration of 25 mg/ml was prepared by dissolving MICO base in a 50/50 (v/v) methanol/50 mM pH 6.8 phosphate buffer solution. The pH of the medium was checked at the end of the experiment.
Stability studies
The stability of the inclusion yield of the complexes produced by SCCO2 was determined. Three combinations were studied: the complexes between MICO base/HPγCD, MICO base/HPγ CD/citric acid and MICO base/HPγCD/L-tartaric acid and the corresponding physical mixtures. The samples were placed in airtight glass containers and stored in a Binder KBF environmental test chamber (Tuttlingen, Germany) at 25°C ± 1°C. All samples were stored in triplicate. The total and free MICO contents were determined in both complexes and physical mixtures after 0, 7, 15, 30, 60, 120, 180, 270 and 360 days of storage.
Results and Discussion
Validation of the differential solubility method
Determination of extraction efficiency
Results of extraction recoveries are presented in table 1.
Table 1: Determination of the extraction efficiency of MICO in physical mixtures with b CD and γ CD.
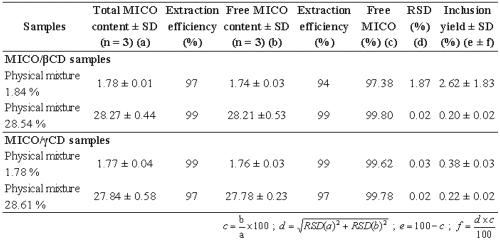
For all physical mixtures, as extraction efficiencies are higher than 94%, the extraction can be considered as complete. The calculated inclusion yields are lower than 2.62%. Since all the MICO is free in physical mixtures, the obtained inclusion values confirm that this differential solubility method is able to solubilize all the free MICO.
Analysis of the precipitated miconazole/CD inclusion complexes
The analysis of reference inclusion complexes between MICO and CD using the differential solubility method and the DSC are given in table 2.
Table 2: Differential solubility analysis and DSC analysis of precipitated MICO inclusion complexes.

In these samples, MICO is completely included in the CD cavity and the free MICO content is equal to zero. The results for the MICO/b CD complex analysis show an inclusion yield of 99.19%, thus confirming the DSC results. For the MICO/ γ CD complex, the measured inclusion yield is 88.67%. The observed difference in the free MICO content between the DSC and the differential solubility method is the result of a pseudopolymorphic transformation of MICO during the preparation of the inclusion complex13. Indeed, MICO can precipitate onto γ CD particles in an amorphous form, which is considered as free MICO by the differential solubility method, but not detected by thermal analysis (DSC). In conclusion, the differential solubility method is able to give an accurate determination of the inclusion yield in MICO/CD complexes.
Effect of both cyclodextrins and acids on the complex formation
Effect of both cyclodextrins and hydroxyacids on the inclusion process
As shown in table 3 for the binary mixtures, no significant inclusion occurs with bCD and HPbCD for either of the two MICO types. The maximum yield, which lies at about 37%, is obtained with the MICO base/HPγ CD combination. Table 3 also shows the influence of five different acids on MICO inclusion (respectively for citric, DL-malic, L-tartaric, maleic and fumaric acids). Compared to binary systems, the yields are remarkably modified by the presence of these acids. Moreover, the use of a hydroxypropylated CD gives better results than those obtained with the native CD. As in the binary complex, the inclusion with b CD is insignificant.
Table 3: MICO inclusion yields in function of MICO, CD and acid types (n = 3).
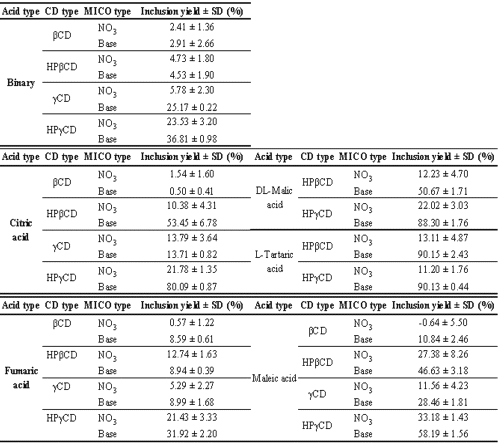
For the ternary mixture containing MICO base, HPγ CD and citric acid, the inclusion yield is close to 80%, while for the same combination with DL-malic or L-tartaric acid, the inclusion is about 90%. These acidic compounds multiply the inclusion yield by two. HPâCD gives similar results. In combination with MICO base and citric or DL-malic acid, the inclusion is close to 50%, whereas L-tartaric acid gives an inclusion yield of around 90%. At this point in the study, several factors that are important for inclusion can be pointed out: the use of MICO base, the presence of both a hydroxyacid (especially L-tartaric acid) and a hydroxypropylated CD, and the CD cavity size. Firstly, in all inclusion experiments, MICO base gives better results than the nitrate does. This could be a result of the diffusion coefficient of MICO in the SCCO2. Indeed, the diffusion coefficient of a guest molecule in the SC medium influences the interactions between the guest molecule and the CD. Data on the diffusion coefficients of MICO base and nitrate in SCCO2 are not available. As SCCO2 presents a poor dielectric constant, ranging from ~ 1 to 2 in function of pressure and temperature14, MICO nitrate should not be dissociated in this medium. Consequently, its diffusion coefficient, which also depends on molecular shape15, should be lower than that of the MICO base, which presents a more compact shape than MICO nitrate's. Moreover, the interactions between the guest and the host are promoted when the guest is not protonated because the polarity of the CD cavity looks like the polarity of a hydro-alcoholic solution16. MICO type also has an effect on the interaction with the acid. Indeed, as mentioned in the literature, the acidic ternary agent interacts with a basic function of the guest and with the hydroxyl group of the CD17. In this case, hydroxyacid does not fit into the CD cavity, but remains outside, while MICO forms an inclusion complex through the interaction between its O-CH2-dichlorobenzene group and the CD cavity18. Electrostatic interactions between the acid and the imidazole ring of the drug take place on one end. Hydrogen bonds with the hydroxyl group of the CD are formed on the other end. So, by bridging the guest and the host, the acid has two interaction points. In the case of MICO nitrate, the imidazole ring is already protonated and cannot interact with the acid by formation of hydrogen bonds. Moreover, the acidic constant of nitric acid (pKa -1.40) is smaller than the pKa of the hydroxyacids, which are not able to shift the nitric acid, thereby limiting the interactions MICO-acid. This is the reason why the yields are not modified very much by the presence of an acidic ternary compound. Secondly, the modified groups on the CD ring have a positive influence on the formation of hydrogen bonds between the acid and the CD. As reported by Zia et al.19, the complexation properties of the CD are modified by the hydroxypropyl substitution. The cavity of the CD seems to be enlarged and new hydrogen bonds can occur between the guest and the host in order to stabilize the formed complex. Lastly, by referring to the solid-state structure of the γCD, the crystallization water included in the cavity is characterized by a high degree of disorder, greater than in the b CD. Some of these water molecules seem to be in an activated state which could promote the formation of inclusion complexes20. This energetically favorable state could explain why inclusion is not possible with the β CD in the supercritical conditions tested.
Effect of the conformation of the ternary compound
In order to estimate the effect of the acid conformation, the influence of fumaric and maleic acid was evaluated. The data are also reported in table 3. These acids increase the inclusion in the same way as the hydroxyacids, but the effect depends on the conformation of the double bond. When the double bond conformation is cis, as for maleic acid, the interactions are higher than when the conformation is trans, like for fumaric acid, except for b CD. So, for the MICO base/HPγ CD/maleic acid complex, the inclusion yield is about 58%, while it is 32% with fumaric acid. This kind of molecular interaction has been described by Muñoz de la Peña et al.21. By forming ternary complexes between b CD, pyrene and several alcohols, they demonstrated that the size and conformation of the alcohol lead to larger equilibrium constant values for the complexes when there is a good matching among the three inclusion complex components.
Differential solubility studies reveal the decrease of MICO in the maleic acid complexes, to the benefit of another compound. It was shown that this compound corresponds to the formation of MICO maleate.
Figure 2a depicts the analysis of total MICO content by differential solubility for a MICO base/HPγ CD/maleic acid complex.
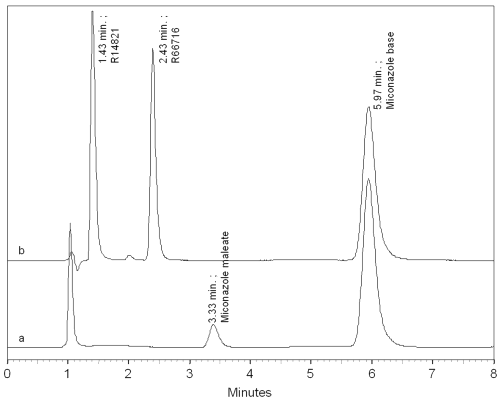
Figure 2: HPLC Chomatogram obtained after the analysis of total MICO content for the MICO base/HPyCD/maleic acid SCCO2 complex (a) and the analysis of R14821, R66716 and MICO base at equal concentration (40 mg/ml) (b).
It shows the presence of MICO base at 5.97 minutes retention time and also the presence of another compound at 3.33 minutes retention time that is not the result of the typical degradation products of MICO base in acidic conditions, i.e. R14821 and R66716. Indeed, the analysis of methanolic solution of R14821, R66716 and MICO base at 40 m g/ml concentration reveals following retention times: 1.43, 2.43 and 5.97 minutes respectively (figure 2b).
A physical mixture of MICO base and maleic acid was prepared in equimolar proportions and left in an oven at 125°C for 60 minutes, these conditions simulating temperature conditions in the SCCO2 experiment. In these circumstances, the MICO base melts (m.p. = 84.3°C) and reacts with maleic acid (m.p. = 139°C) to form MICO maleate. The resulting product shows the same retention time as the product formed after treatment with SCCO2 (data not shown). DSC analyses were performed.
Figure 3 displays the DSC curves of the MICO base, the physical mixture between MICO, and maleic acid and MICO maleate.
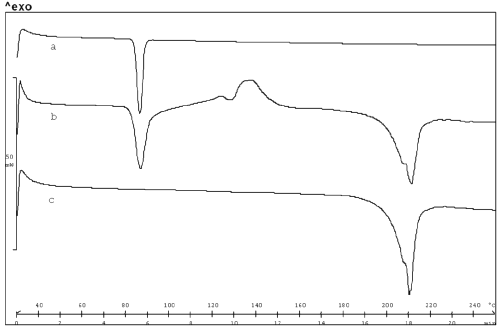
Figure 3: DSC profiles of MICO base (a), physical mixture of MICO base/maleic acid (b) and MICO maleate (c).
The DSC profile of the physical mixture can be divided into different steps: the melting of the MICO base, the melting of maleic acid, an exothermic phenomenon (at 130°C) which is attributed to the salt formation and its crystallization, and finally, the salt melting point at 210°C. Analog thermal behavior has been previously described by Mura et al. with an acidic drug in the presence of a basic aminoacid22. Lastly, LC-MS analysis was performed to confirm the presence of such a salt.
Figure 4 presents the results obtained during the LC-MS analysis of MICO maleate produced as described above. On the first chromatogram (figure 4a), the detection is set at m/z equal to 417 and MICO is observed at the 5.15 minutes retention time. On the second chromatogram (figure 4b), where the detection is set at m/z equal to 533, a product is observed at the retention time of 3.00 minutes. The mass of this product corresponds to the addition of the molecular masses of MICO base (MM = 416) and of maleic acid (MM = 116), which confirms the presence of a MICO base salt: MICO maleate. The chromatogram obtained under base peak ion (BPI) scan mode (figure 4c) confirms the good separation of the compounds.
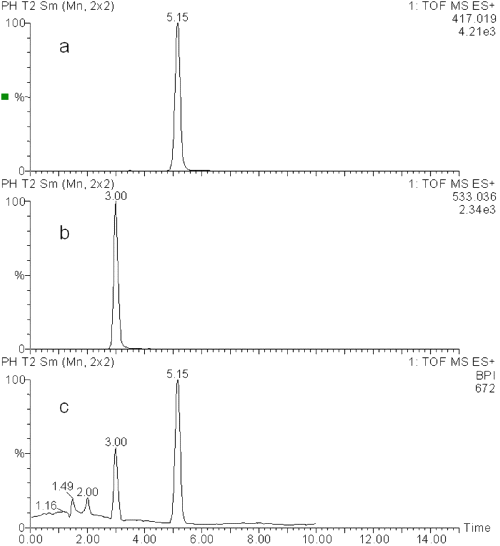
Figure 4: LC-MS chromatograms of a MICO maleate solution: detection set at m/z 417 (a), detection set at m/z 533 (b) and base peak ion (BPI) scan mode (c).
This experiment allows to clearly identifying the compound observed during the analysis of the complexes containing maleic acid by differential solubility (figure 3b).
So the compound detected at the 3.33 minutes retention time was MICO maleate. In conclusion, during the formation of inclusion complexes between CDs, MICO and maleic acid, two phenomena occur. The first is the formation of a ternary complex between the three compounds, and the second is the formation of a salt between MICO base and maleic acid.
Effect of the process time
The formation kinetics of an inclusion complex between MICO and HPγ CD were determined in the presence and in the absence of citric acid. Results are shown in figure 5.
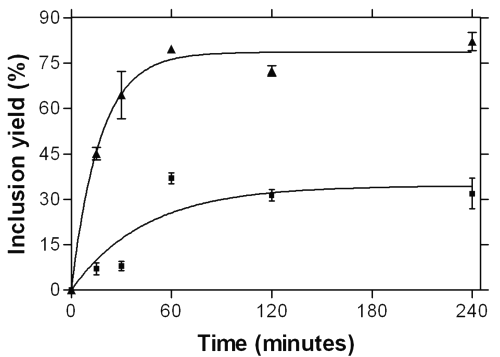
Figure 5: Kinetics of the complex formation by SCCO2 for MICO base into HPγCD (■) and into HPγCD in the presence of citric acid (▲) at 30 MPa and 125°C.
Kinetics profiles of MICO inclusion show a hyperbola shape. In both cases, a plateau is reached after a contact time of 60 minutes. At this time, the inclusion yields are about 37% for the binary mixture and about 81% for the ternary one. So it can be said that the acid does not modify the kinetics of inclusion since the plateau appears at the same contact time, but that it does modify the position of the complexation equilibrium, promoting the formation of the inclusion compound.
Characterization of the complexes
FT-IR spectroscopy
Previous FT-IR analysis has shown that SCCO2 has no influence on the pure compound spectra (data not shown). The typical stretching bands of MICO base were found at 1562 and 1589 cm-1 (dichlorosubstituted benzene) and at 1468, 1506 and 1546 cm-1 (imidazole ring). These bands appear unchanged in the physical mixtures (figure 6).
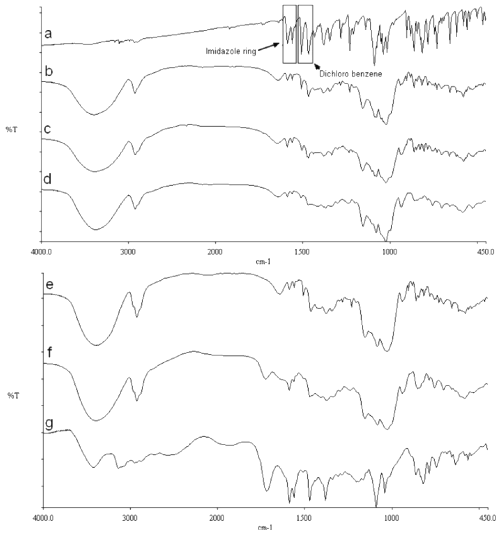
Figure 6: FT-IR spectra of MICO base/CD systems: MICO base (a), physical mixture with γCD (1:1) (b), MICO/γ CD complex produced by SCCO2 processing (c) and MICO/γ CD complex obtained by precipitation (d), MICO base/HPγ CD/citric acid (1:1:1) physical mixture (e), MICO/HPγ CD/citric acid complex produced by SCCO2 processing (f) and MICO/citric acid compound (g).
However, shifts and attenuations of these characteristic bands show that there are some interactions between the MICO and the CD by formation of an inclusion complex (table 4).
Table 4: Characteristic FT-IR bands positions of MICO in several different systems.
The comparison of the FT-IR spectra of the MICO base/HPãCD/citric acid physical mixture (figure 6e), the complex (figure 6f) and the MICO/citric acid compound (figure 6g) produced with the same method as MICO maleate, shows some modifications of the typical MICO bands.
Moreover, a broad absorption band can be observed at 1719 cm-1. This signal, not visible in the physical mixture, is attributed to a carboxylic C=O stretching that generally occurs at 1725 cm-1, while the carboxylate C=O stretching typically occurs at around 1600 cm-1. This signal is amplified by an important interaction between the acid and the imidazole ring of the MICO. This explains why this function is visible in the complex, but not in the corresponding physical mixture. All these observations suggest that MICO interacts with the CD in order to form a genuine inclusion complex, and with the acid, which reinforces the association between the guest and the CD.
Dissolution studies
The dissolution profiles of MICO and of different MICO/CD formulations are depicted in figure 7.
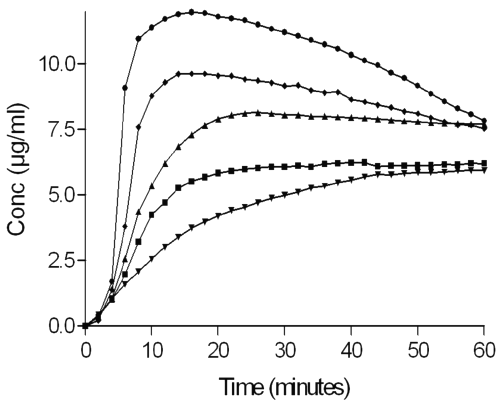
Figure 7: Dissolution curves of MICO base (▼), physical mixture of MICO base/HPγ CD (■), MICO base/HPγ CD complex (▲), physical mixture of MICO base/HPγ CD/L-tartaric acid (◆), MICO base/HPγCD/L-tartaric acid complex (●).
Before comparing the dissolution curves, it is interesting to note that the pH value of the dissolution medium remained unchanged after the different runs. It can be observed that the amount of dissolved MICO from the powders containing cyclodextrins is higher than when the drug is alone. The physical mixtures show evidence of faster dissolution rates than the MICO alone. The complexes give the largest improvement in drug dissolution, particularly the complex containing L-tartaric acid. The modification of the MICO dissolution in the physical mixture is the result of interaction between the drug and the CD by the formation of a soluble inclusion complex in the dissolution medium. Moreover, an acidic micro-pH, favorable to the drug dissolution, is formed thanks to the solubilization of L-tartaric acid. As expected, the complexes formed by SCCO2 processing present better dissolution profiles than the physical mixtures or the MICO alone. The best performance is obtained with the ternary complex between MICO base/HPγCD/L-tartaric acid. This complex already had the highest inclusion yield due to the presence of L-tartaric acid, which seems to promote dissolution. Finally, at the end of the dissolution test, a decrease in the MICO concentration is observed for the ternary complex. This decrease in solubility is a result of the dilution of the complex, which promotes the dissociation of MICO from its inclusion compound, according to the binding constant value. So MICO precipitates to reach the solubility of MICO at the equilibrium in each final medium.
Stability study
The results obtained during the long-term conservation of the complexes are presented in figure 8.
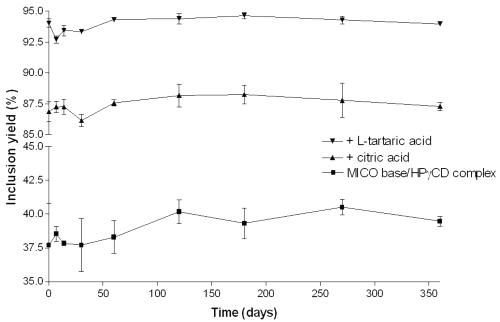
Figure 8: Long-term stability of the MICO base/HPγ CD (■), MICO base/HPγ CD/citric acid (▼) and MICO/HPγ CD/L-tartaric acid (▲) complexes.
In the physical mixtures, no significant inclusion yield occurs (data not shown). Statistical analysis was performed in order to estimate the effect of time on MICO inclusion. The mean values of the inclusion yields for each complex were compared by a one-way variance analysis at the p = 0.05 significance level. The results show that there is no significant modification in the inclusion yield of MICO base for the MICO base/HPγCD complex (p = 0.15), for the MICO base/HPγCD/citric acid complex (p = 0.07) and for the MICO base/HPγCD/L-tartaric acid complex (p = 0.13). So it can be concluded that the complexes are stable during the conservation period tested, i.e. one year.
Conclusions
The study strongly suggests that it is possible to produce genuine inclusion complexes between MICO and CD by SCCO2 processing. Some factors have a positive influence on the inclusion yield, such as the type of CD (CD derivatives and larger cavity size) and the type of MICO (MICO base which possesses a higher diffusion coefficient in SCCO2, in comparison with the nitrate). The addition of an acidic ternary compound dramatically improves inclusion yield. The best results are obtained with the combination of MICO base, HPγCD and DL-malic or L-tartaric acids, and with the combination of MICO base, HPb CD and L-tartaric acid, for which the inclusion reaction is nearly complete. In these ternary complexes, both MICO and CD types influence the drug inclusion, like they do in binary complexes. The study of the kinetics for the MICO base inclusion into HPγCD reveals that the time course of inclusion displays a saturation-type shape curve and that the acidic compound does not modify the kinetics, but rather promotes the inclusion of the drug. Moreover, the conformation of the ternary compound is an important factor because it influences the interaction between guest and host. Stability studies reveal that the complexes indeed present an attractive long-term stability up to one year. During the analysis of the complexes containing MICO base and maleic acid, the presence of a salt, MICO maleate, has been found. It has been characterized by DSC and LC-MS. FT-IR analysis confirmed the existence of genuine MICO/CD inclusion compounds, and the dissolution studies showed that the complexes are more soluble than either the MICO alone or the corresponding physical mixtures. This property could be interesting for use of the complexes in a pharmaceutical dosage form, with a view to improving the drug's bioavailability after oral administration.
Acknowledgements
G. Piel is a postdoctoral researcher supported by the F.N.R.S., Brussels, Belgium.
References
Uekama, K.; Hirayama, F.; Irie, T. Cyclodextrin drug carrier systems. Chem. Rev. 98: 2045-2076, 1998
Loftsson, T.; Fririksdottir, H. The effect of water-soluble polymers on the aqueous solubility and complexing abilities of b-cyclodextrin. Int. J. Pharm. 163: 115-121,1998
Mura, P.; Faucci, M. T.; Manderioli, A.; Bramanti, G. Multicomponent system of econazole with hydroxyacids and cyclodextrins. J. Incl. Phenom. Macro. 39: 131-138,2001
Piel, G.; Pirotte, B.; Delneuville, I.; Neven, P.; Llabres, G.; Delarge, J.; Delattre, L. Study of the influence of both cyclodextrins and L-lysine on the aqueous solubility of nimesulide; isolation and characterization of nimesulide-L-lysine-cyclodextrin complexes. J. Pharm. Sci. 86: 475-480,1997
Van Hees, T.; Piel, G.; Evrard, B.; Otte, X.; Thunus, L.; Delattre, L. Application of supercritical fluid carbon dioxide for the preparation of a piroxicam-b-cyclodextrin inclusion compound. Pharm. Res. 16: 1864-1870,1999
Charoenchaitrakool, M.; Dehghani, F.; Foster, N. R. Utilization of supercritical carbon dioxide for complex formation of ibuprofen and methyl-β-cyclodextrin. Int. J. Pharm. 239: 103-112,2002
Piel, G.; Dive, G.; Evrard, B.; Van Hees, T.; Henry de Hassonville, S.; Delattre, L. Molecular modeling study of b- and g-cyclodextrin complexes with miconazole. Eur. J. Pharm. Sci. 13: 271-279,2001
Pedersen, M. Isolation and antimycotic effect of a genuine miconazole b-cyclodextrin complex. Eur. J. Pharm. Biopharm. 40: 19-23,1994
Jacobsen, J.; Bjerregaard, S.; Pedersen, M. Cyclodextrin inclusion complexes of antimycotics intended to act in the oral cavity - drug supersaturation, toxicity on TR146 cells and release from a delivery system. Eur. J. Pharm. Biopharm. 48: 217-224,1999
Piel, G.; Evrard, B.; Fillet, M.; Llabres, G.; Delattre, L. Development of a non-surfactant parenteral formulation of miconazole by the use of cyclodextrins. Int. J. Pharm. 169: 15-22,1998
Caporal-Gauthier, J.; Nivet, J. M.; Algrande, P. Guide de validation analytique: rapport d'une commission SFSTP. I. Méthodologie. STP Pharma Pratiques , 2: 205-226,1992
Van Hees, T.; Piel, G.; Henry de Hassonville, S.; Evrard, B.; Delattre, L. Determination of the free/included piroxicam ratio in cyclodextrin complexes: comparison between UV spectroscopy and differential scanning calorimetry. Eur. J. Pharm. Sci. 15: 347-353,2002
Pedersen, M.; Pedersen, S.; Sorensen, A. M. Polymorphism of miconazole during preparation of solid systems of the drug and b-cyclodextrins. Pharmaceutica Acta Helvetiae , 68: 43-47,1993
Drake, B.; Smith, R. Measurement of static dielectric constants of supercritical fluid solvents and cosolvents. J. Supercrit. Fluids , 3: 162,1990
Bueno, J. L.; Suarez, J. J.; Dizy, J.; Medina, I. Infinite dilution diffusion coefficients: benzene derivatives as solutes in supercritical Carbon Dioxide. J. Chem. Eng. Data , 38: 344-349,1993
Loftsson, T.; Brewster, M. E. Pharmaceutical Applications of Cyclodextrins. 1. Drug Solubilization and Stabilization. J. Pharm. Sci. 85: 1017-1025,1996
Redenti, E.; Ventura, P.; Fronza, G.; Selva, A.; Rivara, S.; Plazzi, P. V.; Mor, M. Experimental and theoretical analysis of the interaction of (+/-)-cis-ketoconazole with b-cyclodextrin in the presence of (+)-L-tartaric acid. J. Pharm. Sci. 88: 599-607,1999
Piel, G.; Llabres, G.; Evrard, B.; Van Hees, T.; Henry de Hassonville, S.; Delattre, L. A nuclear magnetic resonance study of the miconazole-b-cyclodextrin inclusion complex in an acidic medium: Determination of the structure and stability constants. S. T. P. Pharma Sci. 11: 235-238,2001
Zia, V.; Rajewski, R. A.; Stella, V. J. Effect of cyclodextrin charge on complexation of neutral and charged substrates: comparison of (SBE)7M-bCD to HP-b-CD. Pharm. Res. 18: 667-673,2001
Szejtli, J. Cyclodextrin Technology. Kluwer Academic Publishers: Dordrecht, 1988.
Muños de la Peña, A.; Ndou, T. T.; Zung, J. B.; Greene, K. L.; Live, D. H.; Warner, I. M. Alcohol size as a factor in the ternary complexes formed with pyrene and bcyclodextrin. J. Am. Chem. Soc. 113: 1572-1577,1991
Mura, P.; Maestrelli, F.; Cirri, M. Ternary systems of naproxen with hydroxypropyl-b-cyclodextrin and aminoacids. Int. J. Pharm. 260: 293-302,2003
Corresponding Author: Valéry Barillaro, Laboratory of Pharmaceutical Technology, Department of Pharmacy, University of Liège, CHU-Tour 4, Bat. B36, Avenue de l'Hôpital, 1, 4000 Liège 1, Belgium. v.barillaro@ulg.ac.be
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps