J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 7(3):338-344, 2004
Formulation of sustained - release lithium carbonate matrix tablets: influence of hydrophilic materials on the release rate and in vitro-in vivo evaluation.
Jaber Emami1, Naser Tavakoli
Department of Pharmaceutics;
Ahmad Movahedian
Department of Biochemistry,
Faculty of Pharmacy and Pharmaceutical Sciences, Isfahan University of Medical Sciences, Isfahan, IranReceived 23 March 2004, Revised 9 October 2004, Accepted 15 October 2004, Published 15 November 2004
PDF Version
Abstract
PURPOSE: Conventional lithium carbonate (LC) tablets not only produce rapid and relatively high peak blood levels resulting in adverse effects but also should be administered 3 to 4 times daily. These drawbacks can be overcome by designing a suitable sustained-release LC preparation. Methods: Sustained-release matrix tablets containing 450 mg LC were developed using different types and ratios of polymers including carbopol (CP), Na carboxymethylcellulose (Na CMC) and hydroxypropylmethylcellulose (HPMC). The tablets were prepared by either direct compression or wet granulation. In vitro and in vivo, newly formulated sustained-release LC tablets were compared with sustained-release commercial tablet (Eskalith CR®). In vivo studies were conducted in nine healthy subjects in a cross over design, with a 3´3 Latin square sequence. Pharmacokinetic parameters were estimated using classical methods. Results: The matrix tablets containing 15% CP exhibited suitable release kinetics and uniform absorption characteristics comparable to that of Eskalith CR®. In vivo, this formulation produced a smooth and extended absorption phase very much similar to that of Eskalith CR® with the identical elimination half-life and extent of absorption. Conclusion: In vitro release studies demonstrated that the releases of LC from all formulated sustained matrix tablets were generally sustained. Na CMC, CP, and HPMC can, therefore, be used to modify release rates of LC in hydrophilic matrix tablets. The sustained release behavior of tablets containing 15% CP prevents high peak blood levels in man and could be given twice daily to promote patient compliance during maintenance therapy.
Lithium carbonate (LC) is widely used in the prophylaxis and treatment of manic depression and mania and in the maintenance treatment of recurrent depression. Lithium ion is readily absorbed from the gastrointestinal tract; it is not bound to plasma proteins; its volume of distribution corresponds to 70% of body weight; and elimination takes place through the kidneys, with a half-life of 20 to 24 hours (1-3). The therapeutic index of the drug is narrow (4.2 to 8.3 mg/L) and adverse effects are common even at therapeutic serum lithium concentrations. Long-term therapy has to be adjusted to get serum concentrations between 0.6 and 1.25 mEq/L (4.2 to 8.3 mg/L) (1-5). Conventional LC tablets make the drug immediately available for absorption producing rapid and relatively high peak blood levels resulting in adverse effects associated with high lithium serum concentrations and require frequent daily dosing in maintenance therapy that influence patient compliance (1-5). These drawbacks can be overcome by designing a suitable sustained-release LC preparation. The development of sustained-release or controlled-release formulations of this drug is therefore of therapeutic relevance and has drawn the attention of the pharmaceutical industries. The manufacturing procedures of presently available sustained release LC are generally patented. Probably the simplest and least expensive way to control the release of an active agent is to disperse it in an inert polymeric matrix. In polymeric systems, the active agent is physically blended with the polymer powder and then fused together by compression molding, which is a common process in the pharmaceutical industry (6). Different types of polymers including carbopol (CP), sodium carboxymethylcellulose (Na CMC) and hydroxypropylmethylcellulose (HPMC) have been used to control the release of drug from the dosage forms (7-14).
The objective of the present study was (a) to prepare sustained release LC matrix tablets using hydrophilic matrix materials including CP, Na CMC and HPMC, (b) to examine the in vitro release characteristics of LC from formulated tablets by varying type and composition of matrix blend and (c) to compare the in vivo serum concentration profiles of the adopted sustained-release tablet of LC with a commercially available sustained-release tablet of LC (Eskalith CR®) in healthy subjects.
mATERIALS AND mETHODS
Materials
The following materials were used: LC (E. Merck, Darmstadt, F. R. G), lactose (Fast Flo, Foremost Food Company, San Francisco, Calif. 94104), CP (Carbopol 934P, Goodrich, Zaventem, Belgium), Na CMC (Netherland GO 220), HPMC (K100M, Dow Chemical Company, USA), Microcrystalline cellulose (Avicel® pH 101), Eudragit® (Rhom Pharma, USA), magnesium stearate, and commercially available LC sustained-release tablet, Eskalith CR®.
Methods
In-vitro Studies
Preparation of tablets
The hydrophilic matrix tablets were prepared by either direct compression or wet granulation technique. In the direct compression, sustained-release matrix tablets were formulated to contain 450 mg (69%) of LC, and 15, 10 or 5% of CP or Na CMC of total tablet weight (650 mg). Microcrystalline cellulose was incorporated as filler excipients to maintain the tablet weight constant. Powder were mixed and lubricated with 1% (W/W) magnesium stearate and then directly compressed on a single punch tablet machine (KS 43373-202 Kilian Co, GMBH, Koln-Niehl) at a tablet weight of 650 mg, with a flat, non-beveled punch of 12-mm diameter. Tablet hardness was kept constant within the range of 7-8 kg as measured by an Erweka-TB 24 hardness tester.
In the wet granulation technique, 450 mg LC and HPMC (180, 120, or 60 mg) were granulated with an ethanolic solution of Eudragit® S100 (15.5 mg). Granulates were passed through an 18 mesh screen and dried at 40°C for 2 hours. The dried granulate was mixed with other formulation components, 3.3 mg magnesium stearate and 0.33 mg Aerosil®, and then compressed into flat tablets of 11 mm diameter with a hardness of 6 kg.
Conventional LC tablets were formulated to contain 300 mg LC, 55 mg Na CMC, 200 mg microcrystalline cellulose and 1% magnesium stearate.
Lithium assay
Calibration curves were prepared with human serum or dissolution media spiked with known concentrations of LC. Standard samples were treated the same way as the unknown samples. Quantitation of lithium was accomplished by atomic absorption spectrophotometer (Perkin-Elmer, USA-Norwalk, CT) at the wavelength of 670.8 nm. Within-day and between-day variability of the assay was determined by repeated analysis of quality control samples at concentration ranging from 0.05 to 6 mg/l on the same and three different days.
Dissolution testing
The dissolution of the tablets was performed according to USP 25 specifications Test No. 2 with the paddle method using a dissolution tester (Pharma Test, PTZWS3, Germany). The dissolution medium was 900 ml of distilled water maintained at 37°C with a stirring rate of 100 rpm. At time 0, 0.25, 0.5, 1, 2, 3, 4, 5, 6, 7, and 8 hrs, 3 ml of samples were obtained and an equal volume of medium was added to maintain the volume constant. Samples were filtered, diluted, and analyzed for LC concentration in order to characterize the dissolution profiles.
Kinetic analysis of dissolution data
The drug release data were fitted to the following simple exponential model,
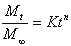
where Mt corresponds to the amount of drug released in time t, M¥ is the total amount of drug released after an infinite time, K is a constant related to the properties of the drug delivery system and n is the release exponent related with drug release mechanism. When n < 0.5, the drug is released from the polymeric matrix with a quasi-Fickian diffusion mechanism, for 0.5<n<1, an anomalous (non-Fickian) drug diffusion occurs and when n >1, a non-Fickian Case II or zero order release kinetics could be observed (15, 16). Mean dissolution time (MDT) was considered as a basis for comparison of the dissolution rates and was estimated by the following equation:
(1)
Where, ABC (area between curves) is the shaded area in Fig 1, Wd (t) is the cumulative amount of drug dissolved at any time interval, and W0 is the actual (as apposed to labeled) quantity of drug, which is available for dissolution.
.gif)
Figure 1: Graphical presentation of the parameters used to estimate the mean dissolution time (MDT).
ABC can be estimated algebraically or arithmetically. In either case, estimation of MDT requires knowledge of the time at which the dissolution process is complete (17). In this study, arithmetic estimation of ABC was made using trapezoidal rule.
In-Vivo Studies
Subjects and blood sampling
The Ethics Committee on human studies of the Isfahan University of Medical Sciences approved the protocol study. Nine healthy adult male subjects aged 21-27 years and weighing 65-85 kg were participated in the single-dose study. The volunteers were informed about the nature and purpose of the experiments for their written consent. Prior to the studies, clinical laboratory tests and physical examinations of all the subjects, including urine cumulative sodium level, serum alkaline phosphatase, urea nitrogen, creatinine, aspartate aminotransferases (AST, SGOT), alanine aminotransferases (ALT, SGPT), and fasting blood sugar were performed to check their health status. Based on medical history, no subjects had a history or evidence of any acute or chronic diseases or drug allergy. All subjects were healthy and free of other drugs for at least one month prior to and during the experiments. Each volunteer was given, in a randomized crossover fashion, based on a 3×3 Latin square sequence, 450 mg LC on three occasions with a one-week washout period. Once as a conventional tablets and twice as a sustained-release tablets, either prepared by direct compression technique in our laboratory or reference standard tablets, Eskalith CR®. The drug was administered with 250 ml of water after overnight fasting. The blood samples were collected for 48 hrs through an indwelling catheter in a forearm vein at 0, 0.5, 1, 2, 3, 4, 5, 6, 8, 12, 24, 36, and 48 hr. The blood samples were allowed to clot and then centrifuged to get serum specimens.
Pharmacokinetic analysis
Pharmacokinetic analysis was performed by means of independent method using MS-Excel software. The maximum LC serum concentration (Cmax) and the corresponding peak time (tmax) were determined by the inspection of the individual drug serum concentration-time profiles. The elimination rate constant (KE) was obtained from the least square fitted terminal log-linear portion of the serum concentration-time profile. The elimination half-life (t1/2) was calculated as 0.693/KE. The area under the curve to the last measurable concentration (AUC 0-t ) was calculated by the linear trapezoidal rule. The area under the curve from zero to infinity (AUC0-¥) was calculated by equation AUC0-t + Ct / KE, where Ct is the last measurable concentration. (18).
The cumulative fraction of drug absorbed at time t was calculated according to the Wagner-Nelson equation (18):
(2)
Where, Ct is the serum LC concentration at time t. The apparent absorption rate constant (Ka) was obtained from the least square fitted log-linear plot of the percent unabsorbed versus time. The absorption half-life (t1/2a) was calculated as 0.693/Ka (18)
Statistical Analysis
One-way analysis of variance (ANOVA) was used to assess the differences between pharmacokinetic parameters of sustained-release formulations with those of conventional formulation and to evaluate the differences between dissolution rate constants and mean dissolution times. Student-t-test was used to compare two means where appropriate. P-value of less than 0.05 was considered significant. The 90% confidence intervals of the ratio of logarithmically transformed pharmacokinetic parameters of optimized sustained-release formulation tablet (CP15%) to reference product (Eskalith CR®) were also estimated. Statistical was performed using SPSS 11.50 for windows.
RESULTS AND DISCUSSION
Linear relationships were found when drug absorbance's were plotted against serum LC concentrations ranging from 0.2 to 6 mg/l (r2 = 0.9995). Coefficient of variation (CV %) and error percent ranged from 10.33 to 3.16% and 10.06 to 1.07%, respectively. The limit of quantitation of the assay was found to be 0.2 mg/l. The data indicate that the method is reproducible within day and between days. Release behaviors of LC from conventional, commercially available (Eskalith CR®) and formulated sustained-release tablets in distilled water are depicted in Fig 2.
.gif)
Figure 2: Release profiles of Lithium carbonate from commercially available tablets and formulated sustained-release matrix tablets (n = 6).
Panel A: Commercially available Eskalith CR® and immediate release formulation (IMR),
Panel B: Matrix tablets using Na carboxymethylcellulose (Na CMC)
Panel C: Matrix tablets using carbopol (CP)
Panel D: Matrix tablets using hydroxypropylmethlcellulose (HPMC).
Each data point represents the mean of six determinations. The drug release kinetics including MDT and release exponents are also presented in Table 1.
Table 1: Mathematical modeling and drug release kinetics of lithium carbonate commercially available and formulated sustained-release tablets.
The release profiles of LC from conventional and presently available sustained-release tablet, Eskalith CR®, are shown in Fig. 2A. As illustrated, 90% of drug was released from conventional tablets within 30 minutes, which is in accordance with USP 25 requirements. However, this time for sustained-release tablet, Eskalith CR®, was, about 6 hours. As shown in Table 1, the MDT for Eskalith CR® is 1.9 ± 0.06 hrs showing more sustained profiles leading to an anomalous mechanism.
As expected, the drug was released much more slowly from tablets containing Na CMC than conventional tablets (Fig. 2B, Table 1). No considerable differences in release rates were observed when 5 and 10 % of Na CMC was incorporated in tablets, however, drug release decreased significantly as 15% Na CMC was used in formulation (MDT, 2.9 ± 0.12, 3.1 ± 0.09, 3.8 ± 0.16, respectively).
As indicated in Fig. 2B, all tablets containing Na CMC showed a relatively slow initial release of drug during the first hour (25-32%), and the release were followed by a sustained manner, which reached 80-85 % of the total content within 8 hours. Na CMC matrices, however, because of its polymer swelling and dissolution properties, did not show initial burst release as observed with HPMC matrices containing 60 mg of the polymer. The release exponent in this series is significantly greater than 0.5, which indicates anomalous drug release. Although the release profiles of this series met the USP specifications, much greater MDT of all tablets containing Na CMC compared to Eskalith CR® indicates much slower release of the drug from this series of formulations. These formulations therefore were not selected for further in vivo studies.
The release patterns of LC from matrix tablet containing CP are illustrated in Fig. 2C. These profiles demonstrate that CP15% has excellent retardant properties when used at 15% level, but less retardant effect at 10 and 5% level (MDT, 2.5 ± 0.09, 1.3 ± 0.04, 0.82 ± 0.02, respectively, Table 1).
As shown in Fig. 2C, when 15% CP was incorporated into formulation, an initial slow release of drug was achieved (10-12 % during the 1st hour). However, the initial slow release pattern in the release profiles of tablets prepared using 5 and 10% CP was not attained. Increasing the amount of CP in the formulations from 5% to 15% resulted in a reduction in the drug release rate (MDT were 0.82 ± 0.02, 1.3 ± 0.04, and 2.50 ± 0.09, respectively), leading to a shift from anomalous type of release towards a swelling-controlled, case II mechanism (Table 1).
This may be due to a reduction in regions of low micro viscosity and the closing of micro pores in the swollen state. Other investigators have observed similar types of results (7, 19). Decreasing content of Na CMC or CP increased the release rate, which also might be due to a decrease in the retardant content and/or to a change in the porosity and tortuosity of the matrix after dissolution of the higher content of water-soluble diluent Avicel®. As Avicel dissolves, it diffuses outward and decreases the tortuosity of the diffusion path of LC (20). It seems that the 15% CP matrix tablets (MDT, 2.5) not only exhibited comparable release kinetics with Eskalith CR® but also meets the USP specifications. This matrix tablet was, therefore, adopted for in vivo studies.
Figure 2D shows the mean dissolution profiles of tablets manufactured using HPMC as matrix material by wet granulation technique.
Although all HPMC matrix tablets demonstrated relatively sustained release behavior, liberation after 4 hr was around 80%. The tablets containing 60 mg of HPMC as compared to those bearing 120 or 180 mg HPMC exhibited a relatively rapid initial release which deviates from USP specifications and differs from Eskalith CR® significantly (MDT, 1.7 ± 0.07, 2.2 ± 0.08, and 2.3 ± 0.11, respectively, Table 1).
The release was identified as anomalous most likely owing to the relative contributions of drug diffusion, polymer relaxation, and matrix erosion to drug release. HPMC matrices containing smaller amount of HPMC showed an initial burst of drug release rate, due to the time required for the formation of an efficient gel layer (16). As the granulating process was conventional, the solution would not form a continuous layer of the acrylic resin over the drug.
Therefore, such structure makes the immediate release of some drug unavoidable. Even small quantity may result in faster initial dissolution of drug. It seems that the matrix tablets containing 120 or 180 mg HPMC (MDT, 2.2 and 2.3, respectively) exhibited comparable release kinetics with Eskalith CR® and meets the USP specifications.
Serum lithium concentrations and standard deviations achieved following oral administration of the conventional, commercially available tablet, Eskalith CR®, and formulated sustained-release tablet, CP15%, are shown graphically in Fig. 3.
Figure 3: Mean serum concentration of lithium (mg/L) following oral administration of 450 mg lithium carbonate in commercially available, Eskalith CR®, and formulated sustained-release matrix tablet CP15% and conventional preparation. (n = 9 volunteers).
The estimated mean values of pharmacokinetic parameters are also listed in Table 2.
Table 2: Pharmacokinetic parameters (Mean ± SD) of various formulations calculated from serum lithium profiles. (n = 9 volunteers).
.gif)
Formulated sustained-release tablets were compared to a standard commercially available sustained-release tablet in order to determine their relative availability and sustained release characteristics. The formulated CP15% matrix sustained-release tablets resulted in Cmax values similar to those produced by the commercially available sustained-release tablets, Eskalith CR® (Table 2, 90% CI = 1.01-1.03). However, higher Cmax was yielded by conventional immediate release formulation (P < 0.05).
As one would expect from inspection of the mean curves, the sustained release tablets had a significantly delayed tmax, showing 6.67 ± 1.03 for the Eskalith CR®, 6.33 ± 1.37 for the CP15% and 2.17 ± 0.41 for the conventional tablets (Table 2). The tmax of the Eskalith CR® and CP15% that showed a smooth and extended absorption phase was significantly longer than that obtained for conventional. This may reduce the incidence of side effects that is usually accompanied with lithium therapy. The differences between AUC values for all tested formulations were not significant (Table 2, P > 0.05, 90% CI=0.989-1.032). The p values for AUC and Cmax were in line with that of the CI. This indicates that the extent of absorption was not different among all formulations. The mean value of the slow disposition rate constants obtained after the administration of the various formulations did not differ. This reflected in the elimination half-lives, which is quite long and did not show statistical differences among various formulations tested (Table 2).
The mean absorption half-lives were found to be 1.60 ± 0.45 hr for standard sustained-release tablets, (Eskalith CR®), 1.21 ± 0.29 for CP15%, and 0.82 ± 0.28 hr for conventional preparation. No differences were observed between mean absorption half-lives of Eskalith CR® and CP15% (P > 0.05). The pharmacokinetic parameters estimated from serum lithium concentrations profiles indicated that CP15% matrix tablets were sustained and exhibited a smooth and extended absorption phase as observed with commercially available sustained-release Eskalith CR®. The initial slow release pattern of the drug observed in vitro in this formulation, will most likely results in a smooth and extended absorption phase of the drug from formulation in vivo. A good correlation between the dissolution profiles and bioavailability was observed. The relationship between percent drug released and percent drug absorbed is illustrated in Fig 4.
.gif)
Figure 4: Relationship between percent lithium carbonate released in vitro and absorbed in vivo.
Conclusion
In vitro release studies demonstrated that the release of LC from all formulated sustained matrix tablets was generally sustained. The drug release from matrices containing Na CMC, CP or HPMC was anomalous while matrices containing 15% of CP or 180 mg of HPMC essentially followed case II release. Therefore, Na CMC, CP, and HPMC can be used to modify release rates of LC in hydrophilic matrix tablets.
The data generated in the present investigation using sustained-release LC tablets indicated that the absorption of LC from gastrointestinal tract may depend mostly on the release rate. A direct correlation between the dissolution profiles of standard Eskalith CR® and CP15% with the relative bioavailability of the formulations could be observed. The sustained release manner of CP 15% may prevent high peak blood levels, wide blood level variations, frequent dosing and gastro-intestinal side effects of LC in man and therefore promotes patient compliance during maintenance therapy. The sustained and controlled release dosage forms of LC have a much slower but continuous absorption as compared to the liquid, tablet, and capsule form. However, the slow-release forms provide equivalent steady-state lithium levels (21).
References
Parfitt, K., Martindale, the complete drug reference, Pharmaceutical Press, 32nd ed., U.S.A., Massachusetts, 1999, p. 290-296.
Baldessarini R. J., and Tarazi F. I. Drugs and the treatment of psychiatric disorders, in: Hardman J. G., Limbird L. E., Goodman and Gilman’s pharmacological basis of therapeutics, 10th ed., McGraw-Hill, USA, 2001, p. 485-520.
Amdisen, A. Serum level monitoring and clinical pharmacokinetics of lithium. Clin Pharmacokin, 2: 73-92. 1994.
Schou, M. Serum lithium monitoring of prophylactic treatment. Critical review and updated recommendations. Clin Pharmacokin, 15: 283-286, 1988.
Carson, S. W. Lithium in: Evans, W. E., Schentag, J. J., Jusko, W. J. (Eds), Applied pharmacokinetics, Principles of therapeutic drug monitoring. 3rd ed. San Franscisco, p. 34 (1-25), 1992.
Lordi, N. Sustained release dosage forms in: Lachman, L., Liberman, H., Kanig, J. (Eds), The theory and practice of industrial pharmacy. Lea & Febiger, Philadelphia, pp. 430-456, 1986.
Khan, G. M., and Zhu, J. B. Studies on drug release kinetics from ibuprofen-carbomer hydrophilic matrix tablets: Influence of co-excipients on release rate of the drug. J Control release, 57: 197-203, 1999.
Huang, L. L., and Schwartz, J. B. Studies on drug release from carbomer tablet matrix. Drug Dev Ind Pharm, 21: 1487-1501, 1995.
Durrani, M. J., Todd, R., Andrews, A., Whitaker, R. W., and Banner, S. C. Studies on drug release kinetics from carbomer matrices. Drug Dev Ind Pharm, 20: 2439-2447, 1994.
Chattaraj, S. C., and Das S. K. Effect of formulation variables on dissolution profiles of diclofenac sodium from ethyl- and hydroxypropylmethylcellulose tablets. Drug Dev Ind Pharm, 22: 555-559, 1996.
Pabon, C.Y., Frutos, P., Lastres, J. L., and Frutos, G. In vitro study of mixed controlled release matrix tablets containing HPMC and polyamide. Drug Dev Ind Pharm, 18: 2163-2171, 1992.
Liu, C.H., Kao, Y. H., Chen, S. C., Sokoloski, T. D., Sheu, M. T., In vitro and in vivo studies of the diclofenac sodium controlled-release matrix tablets. J Pharm Pharmacol, 47: 360-364, 1995.
Lee, B. J., Rayu, S. G., and Cui, J. H. Formulation and release characteristics of hydroxypropylmethylcellulose matrix tablet containing melatonin. Drug Dev Ind Pharm, 25: 493-501, 1999.
Choi, H., Jung, J., Yong, C. S., Rhee, C., Lee, M., Han, J., Park, K., and Kim, C. Formulation and in vivo evaluation of omeperazole buccal adhesive tablet. J Control Release, 68: 405-412, 2000.
Ritger, P. L., and Peppas, N. S. A simple equation for disposition of solute release II: Fickian and anomalous release from swellable devices. J Control Release, 5: 37-42, 1987.
Colombo, P., Bettini, R., Santi, P., Peppas, N. A. Swellable matrices for controlled drug delivery: gel-layer behaviour, mechanisms and optimal performance. Pharm Sci Technol Today, 3: 1-8, 2000.
Macheras, P., Reppas, C., and Dressman, J. B. Biopharmaceutics of orally administered drugs. Ellis Horwood, New York, P 72-73, 1995.
Gibaldi, M., and Perrier, D. Pharmacokinetics. 2nd ed. Marcel Dekker, Inc. New York, p 149-155, 445-449, 1982.
Capan, Y., Ciftci, K., and Hincal, A. A. Influence of filler excipients on the release rate of sustained release lithium carbonate tablets. Eur J Pharm Biopharm, 37: 14-18, 1991.
Ciftci, K., Capan, Y., Ozturk, O., and Hincal, A. A. Formulation and in vitro-in vivo evaluation of sustained release LC tablets. Pharm Res, 7: 359-363, 1990.
Cooper, T. B., Simpson, G. M., Lee, G. H. et al. Evaluation of a slow release lithium carbonate formulation. Am J Psychiatry, 135: 917, 1978.
Corresponding Authors: Jaber Emami, Department of Pharmaceutics, Faculty of Pharmacy and Pharmaceutical Sciences, Isfahan University of Medical Sciences, Isfahan, Iran. emami@pharm.mui.ac.ir
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps
.gif)
.gif)