J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 7(2):252-259, 2004
Tamoxifen in topical liposomes: development, characterization and in-vitro evaluation.
Amit Bhatia
Sun Pharma Advanced Research Centre, Vadodara, Gujrat, IndiaRajiv Kumar, Om Prakash Katare1
University Institute of Pharmaceutical Sciences, Panjab University, Chandigarh, IndiaReceived 9 October 2003, Revised 9 June 2004, Accepted 24 June 2004, Published 16 July 2004
PDF Version
Abstract
Purpose: Tamoxifen, an anti-estrogen compound, has recently been figured as a useful agent in the treatment of certain skin specific disorders. This recent found application has generated an interest in its topical formulation in order to avoid the side effects associated with oral administration, while parenteral administration is restricted due to its limited aqueous solubility. Liposomal carriers, well known for their potential in topical drug delivery, have been chosen to help transport tamoxifen molecules in the skin layers. These vesicles are also expected to provide lipid enriched hydrating conditions to help retain the drug molecules within the dermal layers, at or near to the site of action. With this objective, tamoxifen loaded liposomal systems have been prepared and their topical performance has been compared with non-liposomal systems containing tamoxifen. Method: Multilamellar liposomes of tamoxifen were prepared by thin film hydration method. Various formulation ( viz . lipid composition, drug-lipid ratio, amount and type of surface charge imparting agent etc.) and process parameters (hydration temperature, hydration time etc.) were studied to obtain liposomes with desired attributes. Prepared liposomes were characterized for morphological and micromeritic attributes, employing Malvern mastersizer and optical microscopy. Stability of the liposomes in terms of their drug holding capacity was assessed for a period of 5 weeks, on storage under defined conditions. Liposomal formulations of tamoxifen were evaluated for in-vitro skin permeation, using mice skin. The results thus obtained were compared with that of aqueous solution and Carbopol gel, containing tamoxifen in equal amounts. Results: Optimized process and formulation parameters resulted in multilameller, homogenous population of liposomes in the size range of 1 to 13 mm (mean vesicle diameter 5.3 mm), exhibiting normal size distribution. Maximum loading of tamoxifen was noted to be 57.5% (38.3 m g of drug per mg of lipids), for liposomes composed of hydrogenated phosphatidylcholine and cholesterol, employing 66.6 mg drug per mg of lipids during preparation. Incorporation of dicetylphosphate or stearylamine as charge imparting agent did not influence the vesicular entrapment of TAM in a favorable manner. Amongst different storage conditions, the liposomes stored at 2 to 8°C were found to be most stable, with only 5% drug loss over the storage period of 5 weeks. Significantly higher skin permeation of tamoxifen from liposomal formulations (flux values 63.67 mg/cm2/h and 59.87 mg/cm2/h for liposomal suspension and liposomal gel) has been achieved, as compared to solution (21.65 mg/cm2/h) and Carbopol gel (24.55 mg/cm2/h) containing tamoxifen. Higher magnitude of tamoxifen retention in the skin layers was noted with liposomal formulations vis-à-vis non-liposomal formulations of the drug. Conclusion: Tamoxifen molecules could be successfully entrapped in the liposomes with reasonable drug-loading and desired vesicle specific characters. Higher rate of drug transfer across the skin with liposomal formulations of tamoxifen, suggests that the drug in its lipo-solubilised state might have found facilitated entry into the tough barrier consisting of stratum corneum. The phospholipid enriched amphiphillic nature of the vesicles can be held responsible for modifying the properties of the keratinised layer. Integration of phospholipid molecules with the skin lipids might have served further, to help retain the drug molecules within the skin, thus leading to prolonged presence of drug molecules at the receptor site. These findings have been seen to support the improved and localized drug action in the skin, thus providing a better option to deal with skin-cited problems.
Introduction
Tamoxifen (TAM), an estrogen receptor antagonist is known to be a drug of choice for hormone sensitive breast cancer (1). It has recently been documented to have potential in the treatment of dermatological disorders like psoriasis (2-7). TAM is generally administered through oral and parenteral route. Despite being quite effective on oral administration, TAM exhibits certain side effects like distaste for food, abdominal cramps, nausea and vomiting. However, its other infrequent side effects include endometrial carcinoma, ocular problems, thromboembolic disorders and acquired drug resistance on long-term therapy (8-10). The problems associated with oral administration of TAM, along with difficulty in parenteral administration owing to its limited (0.5mg/ml) aqueous solubility, led the researchers to explore other alternative routes of administration. Enhanced transdermal delivery of TAM, employing different penetration enhancers has been reported (11-12). Topical administration of TAM has recently been found to be effective in the treatment of excessive dermal scarring (5). In another study on topical application of TAM employing different melanoma models, percutaneous administration of TAM yielded higher local tissue concentrations with minimal systemic absorption (13). Similarly, Soe et al. (1997) evaluated the therapeutic advantage with percutaneous application of TAM for the treatment of tumors (14). Significantly high local (subcutaneous and skin) concentration of the drug has been achieved, with lesser drug distribution to other organs. The authors suggested the potential role of topical application, to enhance the therapeutic effect of TAM in the treatment of cancer. Besides TAM, percutaneous application of 4-hydroxy tamoxifen, an active metabolite of TAM, has also been found to exhibit anticancer activity (15-17). However, despite such studies for the topical applications of TAM and its potential in deep-seated dermatological disorders, no topical dosage form of TAM has so far been developed. The present scenario is full of opportunities in this regard, as there has been a substantial progress in the design and development of topical carriers. Amongst the many, phospholipid-based vesicles, i.e., liposomes have been shown to possess a significant potential.
Liposomes are microscopic structures consisting of one or more concentric spheres of lipid bilayers, enclosing aqueous compartments (18). As drug carrier systems for topical treatment, liposomes have been noted to be superior over conventional topical preparations. Phospholipids, being the major component of liposomal systems, can easily get integrated with the skin lipids and maintain the desired hydration conditions to improve drug penetration and localization in the skin layers (19-21). Thus, recognizing the need for topical delivery of TAM and the promising potential of liposomes, it has been envisaged to entrap the drug into these carriers. Interestingly, the incorporation of TAM into lipid bilayers has been viewed to bring additional benefit of imparting stability to the liposomes. The latter is related to the cholesterol like structure of TAM, which on its incorporation in liposomes reduces the flux of molecules. TAM also inhibits the lipid per-oxidation, besides its ability to influence the fluidity of liposomal bilayers in a dose-dependent manner (22-27).
The current study includes the preparation of TAM-loaded liposomes by investigating the influence of different formulation and process related variables. Various parameters viz . percent drug loading (PDL), vesicular size distribution and drug-leakage profile has been assessed. Percutaneous permeation experiments using mice skin have been carried out in-vitro , to investigate the plausibility of delivery of TAM using topical route.
Materials and Methods
Materials
Tamoxifen citrate (Biochem Pharmaceutical Industries, India) and saturated soy lecithin (PC; PhospholiponÒ 90H with phosphatidylcholine content ³ 97%, Nattermann Phospholipids GmbH, Germany) were provided ex-gratis by the respective sources. Cholesterol (CHOL), Sephadex G-50 medium (bead size range 50-150 mm), Dicetyl phosphate (DCP) and Stearylamine (SA) were procured from Sigma Chemical Co. (St. Louis, MO, USA). All other ingredients used in the study were of analytical grade. Double-distilled water was used throughout the experiments.
Method
Multilamellar liposomes were prepared employing thin film hydration technique (28). A lipid phase was prepared by dissolving accurately weighed quantities of the drug, PC and CHOL (Table 1) in the chloroform-methanol mixture (2:1, v/v), in 250 ml round-bottom flask containing glass beads.
Table 1: Effect of drug-lipid ratio and amount of cholesterol on the TAM loading in PC liposomes.
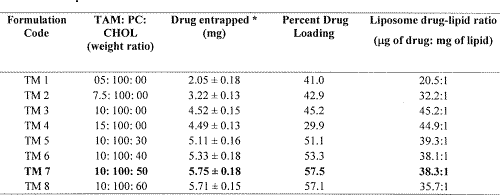
*Value indicates mean ± S.D. (n=3)
The solvent mixture was removed from the lipid phase by rotary evaporation at 45-50°C (Buchi RE 121 Rotavapour, Buchi Laboratories AG, Flawil/Sheiz, Switzerland), to obtain a thin film of lipids on the wall of the flask and the surface of beads. Subsequently, the flask was kept overnight under vacuum to ensure the complete removal of residual solvent. The dry lipid film was hydrated with saline solution at a temperature of 60±2°C. The dispersion thus obtained was vortexed for about 2 minutes. The dispersion was left undisturbed at room temperature for 2-3 hours to allow complete swelling of the lipid film and hence to obtain vesicular suspension. Liposomes, containing varied amounts of charge imparting agent, i.e., DCP or SA, were also prepared employing the same technique.
Drug entrapment studies
Separation of unentrapped drug from the prepared liposomes was carried out by mini column centrifugation method (29-30). Liposomal suspension (0.2 ml) was placed in Sephadex G-50 column (pre-saturated with empty liposomes) and centrifuged at 2000 rpm for 3 min. Elutes containing drug loaded liposomes were collected and observed under optical microscope to ensure the absence of unentrapped drug particles. Appropriate amount of elute was digested with chloroform-methanol (2:1, v/v) and the clear solution thus obtained was analyzed spectrophotometerically (U.V./Visible spectrophotometer, Shimadzu-1601, India) for the drug content estimation at a lmax of 274 nm. Liposomes prepared without drug were treated in similar manner and served as blank for the above study. Studies were conducted in triplicate. Percent drug loading (PDL) for the prepared liposomes was calculated as in Eqn 1.
(1)
Microscopy and size distribution profile
Prepared liposomal batches were monitored for their morphological attributes using optical microscope (at suitable magnification). Size distribution profile of liposomes was determined by light scattering based on laser diffraction method employing Malvern Mastersizer (Model-S, version 2.15, Malvern, UK).
Storage - stability studies
The ability of vesicles to retain the drug (i.e., drug-retentive behavior) was assessed by keeping the liposomal suspensions at four different temperature conditions, i.e., 4-8°C (Refrigerator; RF), 25±2°C (Room temperature; RT), 37±2°C and 45±2°C for a period of 5 weeks (31). The liposomal suspensions were kept in sealed ampoules (10ml capacity) after flushing with nitrogen. Samples were withdrawn periodically and analyzed for the drug content, in the manner described under drug entrapment studies.
In-vitro skin permeation studies
Skin permeation studies with TAM-containing liposomal formulations (liposome suspension and liposomes incorporated in Carbopol gel), were carried out using abdominal skin of LACA mice, employing modified Franz-diffusion cells. The results obtained were compared with that of non-liposomal formulations of TAM, i.e., aqueous solution and Carbopol gel, containing equivalent amounts of TAM. Briefly, to obtain skin, animals were sacrificed after getting approval of the animal ethics committee. Hair on the dorsal side of the animal were removed with the help of a 0.1 mm animal hair clipper, in the direction of tail to head. Dermis part of the skin was wiped 3 to 4 times with a wet cotton swab soaked in isopropanol to remove any adhering fat material. Skin was mounted on the receptor chamber with cross-sectional area of 3.801 cm 2 exposed to the receptor compartment. Saline solution (35 ml, 0.9% w/v) was employed as receptor phase and the temperature was maintained at 37±2°C. Liposomal or non-liposomal TAM formulation (amount equivalent to 3 mg of drug) was applied uniformly on the dorsal side of mice skin. Aliquots of 3 ml were withdrawn periodically and replaced with same amount of saline solution to maintain the receptor phase volume at a constant level. The samples were quantified spectrophotometerically at a lmax of 274 nm.
Determination of TAM retention in skin
The ability of vesicles to help retain the drug within the skin milieu (i.e., depot-effect) was investigated by determining the amount of drug retained in the skin samples employed in permeation studies. After completion of the permeation experiment, skin mounted on the diffusion cell was removed. The skin was cleaned with cotton dipped in saline solution and blotted with tissue paper to remove any adhering formulation. Subsequently, the skin sample was homogenized with 10 ml of chloroform:methanol mixture (2:1, v/v), for the extraction of TAM. Homogenate suspension thus obtained was filtered using membrane filter (0.45m) and quantified for the drug content.
Results
Preparation of drug loaded liposomes
Various product-influencing variables viz. vacuum, speed of rotation, hydration media and hydration time were studied in order to produce TAM loaded liposomes with desired attributes. Rotational speed of the flask demonstrated marked influence on the thickness and uniformity of the lipid film. The optimum speed was noted to be 150 rpm, as the same yielded a uniform thin film on the flask and subsequently homogeneous population of liposomes. The film was kept under vacuum overnight to achieve complete drying and hence, to avoid the formation of emulsion. The latter may result due to the presence of residual organic solvent in the lipid film during hydration. Two-minute vortexing was found to be appropriate to obtain the liposomal suspension free from aggregates. The process of vortexing did not affect the percent entrapment of drug in liposomes, which was confirmed by determining drug entrapment in liposomes, before and after vortexing. With regard to the influence of formulation components on the PDL, different compositions with varying ratios of drug, PC and CHOL (with or without DCP or SA) were studied. Table 1 summarizes the influence of drug-lipid ratio and the effect of CHOL on the PDL of TAM in the liposomes. In case of CHOL free soy PC liposomes (formulation TM 1 to TM 4), maximum drug loading of 45.2 percent (i.e., 45.2 m g drug per mg of lipids) could be achieved using 1:10 w/w drug-lipid ratio during preparation. A further improvement of 5 to 12 % in the entrapment of TM 3 liposomes was noted with the addition of 30 to 50% w/w of CHOL (formulation TM 5 to TM 7, Table 1). Albeit the PDL was found to increase with CHOL addition, however, the effective drug-lipid ratio in the liposomes decreased due to increase in the total amount of lipids. Surface charge imparting agents, i.e., DCP and SA were also investigated individually for their effect on PDL in the TM 7 liposomes. It was observed that the inclusion of DCP or SA did not affect the vesicular entrapment of TAM (data not shown) in a favorable manner. Based on the above findings, liposomal formulation TM 7 was selected for further studies.
Characterization and stability profile of liposomes
The size range of TM 7 liposomes was found to be 1 to 13 mm, with 90% of the liposomal population equal or below 12.9 mm. The mean vesicle diameter was found to be 5.3 m m. Log-size distribution curve (Figure 1) confirms the normal size distribution of the vesicles.
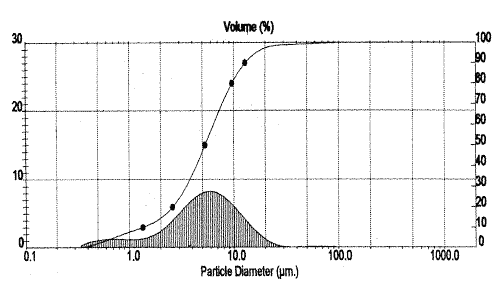
Figure 1: Particle size distribution profile of liposomal formulation (TM 7) of tamoxifen.
The reproducibility of the liposomal formulation with respect to size was confirmed by preparing the formulation six times, but the statistical analysis was avoided, as the particle size data was highly reproducible each time. The Optical photomicrograph of TM 7 liposomes (Figure 2) obtained at suitable magnification (1000 x) confirms the multilamellar nature of the vesicles.
Figure 2: Optical photomicrograph of formulated liposomes loaded with tamoxifen.
TM 7 liposomes were found to be reasonably stable in terms of aggregation, fusion and/or vesicle disruption tendencies, over the studied storage period. Liposomes also retained their multilamellar nature and shape uniformity to an appreciable extent. The bar diagram (Figure 3) depicts the percent drug leakage from liposomes over the 5 weeks period, at different storage temperatures.
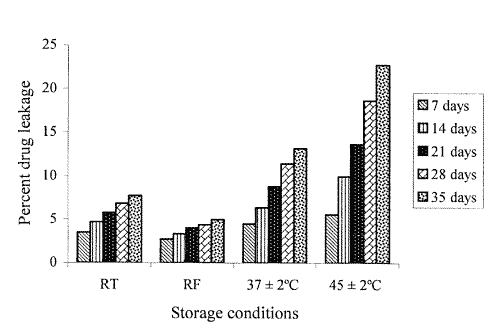
Figure 3: Extent of drug leakage from TM 7 liposomes at different storage temperatures.
Substantial loss of drug, i.e., approximately 14 % to 22 % was evident from the samples stored at elevated temperatures, i.e., 37±2°C and 45±2°C, respectively. On the other hand, at lower temperature conditions, i.e., RT and RF, the liposomes could retain 93 % and 95 % of the incorporated drug, respectively.
Skin permeation and skin retention studies
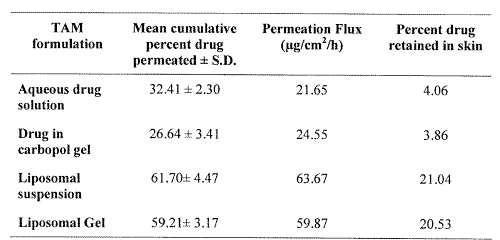
Results obtained from in-vitro drug permeation studies conducted with different formulations of TAM, are shown in Table 2.
Table 2: In-vitro skin permeation and skin retention of tamoxifen from different formulations.
Significant augmentation in the skin permeation of TAM has been observed (Figure 4) with liposomal formulations vis-à-vis aqueous solution or Carbopol gel.
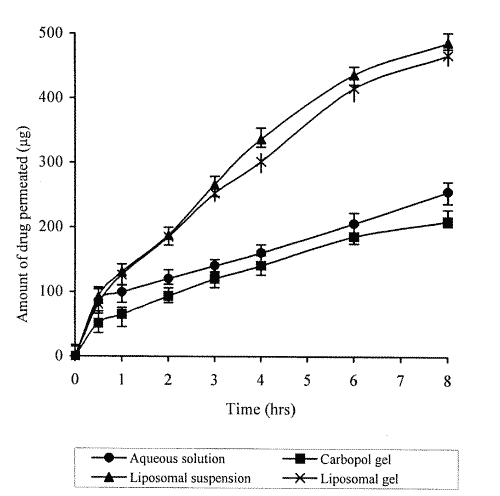
Figure 4: Permeation profile of different tamoxifen containing systems across mouse skin.
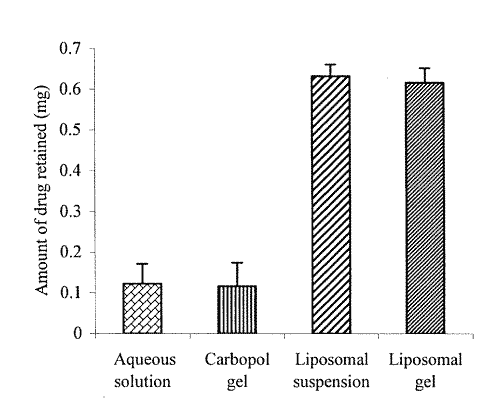
The amount of TAM permeated in eight hrs was found to be 61.7 % and 59.2 % from liposomal suspension and liposomal gel, respectively, whereas only 32 % and 27 % of the drug permeated in case of aqueous solution and Carbopol gel, respectively. Higher values of flux obtained with liposomal suspension (63.67 mg/cm2/h), and liposomal gel (59.87 mg/cm2/h), than that obtained with aqueous solution (21.65 mg/cm2/h) and Carbopol gel (24.55 mg/cm2/h), clearly vouch for the permeation enhancing effect of vesiculation on the drug. The bar diagram (Figure 5) indicates the amount of TAM retained in skin with its different formulations.
Figure 5: Amount of tamoxifen retained in skin with different formulations.
Results of this study clearly depict that the amount of drug retained in the skin was considerably higher in case of liposomal preparations, than with non-liposomal formulations. This gave an understanding that liposomes could not only enhance the penetration of drug molecules but also helped localize the drug in the skin.
Discussion
Formulation aspects
Tamoxifen was chosen for encapsulation within the phospholipid bound closed lamellar systems in order to explore its potential for topical application. The encapsulated molecules of the active substance, in the lipoidal polar-nonpolar folds of liposomes were expected to be delivered into the skin layers, as desired.
The preparation of TAM loaded liposomes was initiated by studying the influence of drug-lipid ratio on drug entrapment in vesicles. The drug bearing capacity of liposomes (i.e., PDL) was found to be invariably dependent on drug-lipid ratio employed in the liposomal composition. A considerable enhancement in the PDL values was observed with increase in the amount of TAM from 5 mg to 10 mg, i.e., upto drug-lipid ratio of 1:10 w/w (while keeping the amount of lipids fixed, i.e., 100 mg). However, the values of PDL tended to decrease with further increase in amount of the drug. The same may be ascribed to the saturation of liposomal lipid domains with reference to drug, wherein the net decrease in the PC content provides limited entrapment capacity (32). Incorporation of CHOL enhanced the percent entrapment of TAM, owing to its cementing effect on the membrane packing. The same would prevent drug leakage from the bilayer membranes leading to enhanced drug retention in liposomes (33). Use of charge imparting agent, i.e., DCP or SA, did not influence the entrapment of TAM in the liposomal compartments. This observation is quite contrary to the earlier reports (32, 34), indicating higher drug entrapment in the vesicles containing DCP or SA, which is usually attributed to improved structural integrity of the vesicles.
Characterization and stability profile of liposomes
The magnitude of drug retention within the vesicles, on storage under defined conditions, ultimately governs the shelf life of the developed formulation. Acceleration in drug leakage at higher temperatures, as observed in storage-stability studies, suggested keeping the liposomal product in the refrigeration conditions, to minimize the drug leakage from liposomal-systems. Loss of drug from the vesicles stored at elevated temperatures may be attributed to the effect of temperature on the gel to liquid transition of lipid bilayers together with possible chemical degradation of the phospholipids, leading to defects in the membrane packing (31-33). The drug leakage of less than 5% of the initial load at refrigeration conditions is well within the limits, when vesicles are to be advocated for topical applications.
In-vitro performance of liposomes
Improved skin permeation of TAM coupled with its enhanced retention in the skin with liposomal formulation can be ascribed to the lipo-solublized state of TAM molecules. The latter was achieved in the presence of aqueous and non-aqueous phase of bilayered systems, a state most ideally suited for drug penetration. The liposomal phospholipids (also one of the natural constituent of skin lipids) helped generating and retaining the required physico-chemical state of the skin for enhanced permeation of the TAM. Further, the phospholipid-rich domains of vesicles might have helped to produce the depot effect for drug molecules. The latter has been reflected as higher amount of drug retention within the skin layers in case of liposomal formulations.
Thus, the liposomal TAM formulation, with desired characteristics for topical administration, could be successfully prepared. The formulated TAM liposomes have shown an appreciably enhanced skin permeation as well as retention of drug molecules in the skin. These results advocate the extension of this work on a collaborated platform. The preliminary clinical trials of the developed liposomal formulation are underway at the Dermatology department of Post Graduate Institute of Medical Education and Research, Chandigarh, India.
Acknowledgments
We are thankful to Biochem Pharmaceutical Industries, Mumbai, India and Nattermann Phosphopids GmbH Germany, for generously providing the Gift samples of Tamoxifen and Phospholipon® 90 H, respectively. Our thanks are also due to University Grants Commission, New Delhi, India, for providing financial assistance for carrying out the research work.
References
Clarke. R., Liu, M. C. and Bouker, K. B., Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene, 22: 7316-7339, 2003.
Cohen, M. H., Hirschfeld. S., Flamm Honig, S., Ibrahim, A., Johnson, J. R., O’Leary, J. J., White, R. M., Williams, G. A. and Pazdur, R., Drug approval summaries: arsenic trioxide, tamoxifen citrate, anastrazole, paclitaxel, bexarotene. Oncologist, 6: 4-11, 2001.
Stevens, H. P., Ostlere, L. S., Black, C. M., Jacobs, H. S. and Rustin, M. H., Cyclical psoriatic arthritis responding to anti-oestrogen therapy. Br J Dermatol, 129: 458-460, 1993.
Ferrari, V. D. and Jirillo, A., Psoriasis and tamoxifen therapy: a case report. Tumori, 82: 262-263, 1996.
Hu, D., Hughes, M. A. and Cherry, G. W., Topical tamoxifen-a potential therapeutic regime in treating excessive dermal scarring? Br J Plast Surg, 5: 462-469, 1998.
Boyd, A. S. and King, L. E. Jr., Tamoxifen induced remission of psoriasis. J Am Acad Dermatol, 41: 887-889, 1999.
Shah, M. G. and Maibach, H. I., Estrogen and skin. An overview. Am J Clin Dermatol, 2: 143-150, 2001.
Morrow, M. and Jordan, V. C., Risk factors and the prevention of breast cancer with tamoxifen. In Cancer Surveys: Breast cancer, ed. JT Papademitrio, I Fentiman, Imperial Res. Fund. London: Cold Spring Harbor Lab. Press, 18: 211-229, 1993.
Jordan, V. C., Tamoxifen: toxicities and drug resistance during the treatment and prevention of breast cancer. Annu Rev Pharmacol Toxicol, 35: 195-211, 1995.
Brigger, I., Chaminade, P., Marsaud, V., Appel, M., Besnard, M., Gurny, R., Renoir, M. and Couvreur, P., Tamoxifen encapsulation within polyethylene glycol-coated nanospheres. A new antiestrogen formulation. Int J Pharm, 214: 37-42, 2001.
Zhao, K., Singh, S. and Singh, J., Effect of menthone on the in-vitro percutaneous absorption of tamoxifen and skin reversibility. Int J Pharm, 219: 177-181, 2001.
El-Kattan, A. F., Asbill, C. S., Kim, N. and Michniak, B. B., The effect of terpene enhancers on the percutaneous permeation of drugs with different lipophilicities. Int J Pharm, 215: 229-240, 2001.
Maenpaa, J., Dooley, T., Wurz, G., VandeBerg, J., Robinson, E., Emshoff, V., Sipila, P., Wiebe, V., Day, C. and DeGregorio, M., Topical toremifine: a new approach for cutaneous melanoma. Cancer Chemother Pharmacol, 32: 392-395, 1993.
Soe, L., Wurz, G. T., Maenpaa, J. U., Hubbard, G. B., Cadman, T. B., Wiebe, V. J., Theon, A. P. and DeGregorio, M. W., Tissue distribution of transdermal toremifene. Cancer Chemother Pharmacol, 39: 513-520, 1997.
Kuttenn, F. and Mauvais-Jarvis, P., Intratumoral levels and metabolism of 4-hydroxytamoxifen after percutaneous administration at the breast level. C R Acad Sci III, 300: 457-462, 1985.
Mauvais-Jarvis, P., Baudot, N., Castaigne, D., Banzet, P. and Kuttenn, F., trans-4-Hydroxytamoxifen concentration and metabolism after local percutaneous administration to human breast. Cancer Res, 46: 1521-1525, 1986.
Sauvez, F., Drouin, D. S., Attia, M., Bertheux, H. and Forster, R., Cutaneously applied 4-hydroxytamoxifen is not carcinogenic in female rats. Carcinogenesis, 20: 843-850, 1999.
Bangham, A. D., Standish, M. M. and Watkins, J. C., Diffusion of univalent ions across the swollen phospholipids. J Mol Biol, 13: 238-252, 1965.
Moghimi, S. M. and Patel, H. M., Current progress and future prospects of liposomes in dermal drug delivery. J Microencapsul, 10: 155-62, 1993.
Cevc, G., Transferosomes, liposomes and other lipid suspensions on the skin: permeation enhancement, vesicle penetration and transdermal drug delivery. Crit Rev Ther Drug Carrier Syst, 13: 257-388, 1996.
Schmid, M. H. and Korting, H. C., Therapeutic progress with liposome drugs for skin disease. Adv Drug Deliv Rev, 18: 335-342, 1996.
Wiseman, H., Cannon, M., Arnstein, H. R. and Halliwell, B., Mechanism of inhibition of lipid peroxidation by tamoxifen and 4-hydroxytamoxifen introduced into liposomes. Similarity to cholestrol and ergosterol. FEBS Lett, 274: 107-110, 1990.
Wiseman, H., Quinn, P. and Halliwell, B., Tamoxifen and related compounds decrease membrane fluidity in liposomes. Mechanism for the antioxidant action of tamoxifen and relevance to its anticancer and cardioprotective actions. FEBS Lett, 330: 53-56, 1993.
Custodio, J. B., Almeida, L. M. and Madeira, V. M., The anticancer drug tamoxifen induces changes in the physical properties of the model and native membranes. Biochim Biophys Acta, 1150: 123-129, 1993.
Wiseman, H., Quinn, P. and Halliwell, B., Tamoxifen and related compounds decrease membrane fluidity in liposomes. Mechanism for the antioxidant action of tamoxifen and relevance to its anticancer and cardioprotective actions. FEBS Lett, 330: 53-56, 1993.
Kayyali, R., Marriott, C. and Wiseman, H., Tamoxifen decreases drug efflux from liposomes: relevance to its ability to reverse multidrug resistance in cancer cells? FEBS Lett, 344: 221-224, 1994.
Severcan, F., Kazanci, N. and Zorlu, F., Tamoxifen increases membrane fluidity at high concentrations. Biosci Rep, 20: 177-184, 2000.
New, R. R. C., Liposomes: A Practical Approach. IRC, Oxford, pp 256 – 258, 1990.
Dipali, S. R., Kulkarni, S. B. and Betagiri, G. V., Comparative study of separation of non-encapsulated drug from unilamellar liposomes by various methods. J Pharm Pharmacol, 48 (11): 1112-1115, 1996.
Sorensen, E. N., Weisman, G. and Vidaver, G. A., A sephadex column for measuring uptake and loss of low molecular weight solutes from small vesicles. Anal Biochem, 82: 376-384, 1977.
Tiwari, S. B., Udupa, N., Rao, B. S. S. and Puma, D., Thermosensiitive liposomes and localized hyperthermia–an effective bimodality approach for tumour management. Int J Pharm, 32: 214-220, 2000.
Aggarwal, R., Katare, O. P. and Vyas, S. P., Prepration and in-vitro evaluation of liposomal/niosomal delivery systems for antipsoriatic drug dithranol. Int J Pharm, 228: 43-52, 2001.
Aggarwal, R. and Katare, O. P., Miconazole nitrate–loaded topical liposomes. Pharm Tech, 26: 48-60, 2002.
Mohammed, A. R., Coombes, A. G. A., Fitzgerald, M. and Perrie, Y., Liposme incorporation of hydrophobic drugs: the effect of charged lipid surfactant. J Pharm Pharmacol, 54 (supplement): S-2, 2002.
Corresponding Author: O. P. Katare, Reader in Pharmaceutics, University Institute of Pharmaceutical Sciences, Panjab University Campus, Chandigarh, India. drkatare@yahoo.com
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps