J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 7(2):217-226, 2004
Cyclooxygenase-3: axiom, dogma, anomaly, enigma or splice error? - not as easy as 1, 2, 3.
Neal M. Davies1, Renee L. Good, Kathryn A. Roupe, Jaime A. Yáñez
College of Pharmacy, Department of Pharmaceutical Sciences and Pharmacology and Toxicology Graduate Program, Washington State University, Pullman, Washington, USAReceived 30 June 2004, Revised 1 July 2004, Accepted 2 July 2004, Published 9 July 2004
PDF Version
Abstract
A continued need to develop safe and effective analgesics and anti-inflammatory drugs fuels the ongoing investigations of cyclooxygenase (COX). In addition, a long unanswered question in the biomedical arena revolves around the mechanism of action of acetaminophen leading to its analgesic and antipyretic activity. Upon the discovery of COX in 1971, alternative enzyme forms were initially suggested. With the development of molecular biology, the discovery of two major cyclooxygenase genes (COX-1 and COX-2) was heralded in 1990, which has subsequently led to the clinical use and development of selective COX-2 inhibitors. Splice variants of both COX-1 and COX-2 were first encountered in the early 1990s, as were single nucleotide polymorphisms of COX-1 and 2. There have been some recently well-publicized investigations of COX-1 and -2 enzyme variants that may assist in our eventual conceptual understanding of the mechanisms of action of acetaminophen. The term "COX-3" has been utilized by some scientists and in the media. The evidence for "COX-3" in terms of various COX-variants and possible scientific and therapeutic implications of COX variant enzymes are described.
Introduction
Over thirty years ago, the mechanism of action of aspirin-like drugs or non-steroidal anti-inflammatory drugs (NSAIDs) was proposed through their inhibition of prostaglandin biosynthesis via the enzyme cyclooxygenase (COX or prostaglandin H2 synthase) (1). In 1982, Sir John Vane's efforts were recognized with a Nobel Prize in Medicine. Some 20 years after the initial discovery it was discovered that there are at least two COX isoforms, COX-1 and COX-2 in 1990 (2). Current research has focused on developing safer NSAIDs based on a better understanding of the existence of the two COX isoforms and their mechanisms of action and physiological roles in the pathogenesis of inflammation.
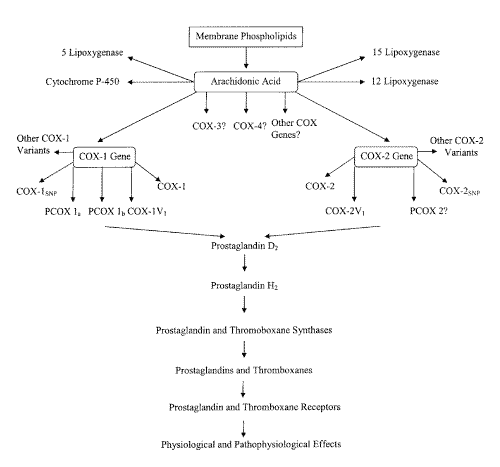
Figure 1: Schematic of Pathways of Prostaglandin and Thromboxane Formation and Action.
New classes of selective COX-2 inhibiting medications have entered the worldwide market based on our increased understanding of COX inhibition. These new medications are safer alternatives to the current NSAIDs in terms of gastrointestinal safety; however, toxicological concerns regarding their renal and cardiovascular safety remain (3). As well as benefiting arthritic patients, these specific inhibitors of COX-2 may demonstrate new therapeutic potential, slowing down tumor growth, delaying the birth process, and impeding the degenerative changes associated with Alzheimer's disease (3).
Almost all available non-specific NSAIDs block both COX isoforms, which can decrease the amounts of prostaglandins formed by COX-1 and -2. There has been much speculation on the part of scientists over the years (4), and some recent research that suggests the possibility of a third COX isoform with the cognomen "COX-3" (5-6).
HOW DOES ACETAMINOPHIN/PARACETAMOL WORK?
Acetaminophen/paracetamol was first used in medicine in 1893 and gained popularity since 1949 after demonstrating pain and fever-relieving properties. Tylenol® is the common brand name in North America. Acetaminophen is a poor inhibitor of COX-1 and COX-2, yet it remains a well-used analgesic/anti-pyretic by millions of people. It has been puzzling to pharmaceutical and medical science that acetaminophen relieves pain and fever; however, it is not a particularly effective anti-inflammatory drug.
Some thirty years ago, using dog brains as a source of enzymes Flower and Vane (4) suggested that acetaminophen acted centrally by inhibiting a COX enzyme. Some studies with COX isozymes as well as studies in homogenates or isolated microsomes have shown that COX-1 and COX-2 are quite insensitive to acetaminophen (7). Other studies suggest acetaminophen has been shown to inhibit activity of COX-1 and COX-2 in isolated tissues and cells, and to reduce prostaglandin production in vivo (8). More recently, Graham and Scott (9) have provided a thought provoking review of this subject. Acetaminophen is a weak inhibitor of COX in isolated enzymes and is a potent inhibitor of prostaglandins in intact cells when low concentrations of arachidonic acid or following activation of the delayed pathway via cytokines to produce prostaglandins (9). The inhibitory concentrations of acetaminophen under these conditions are within the therapeutic range of plasma concentrations ~10 m M. Effects of acetaminophen are not as apparent when arachidonic acid is greater than ~5 m M and when intermediate hydroperoxides levels are increased. It has been suggested that some preparations used for measuring COX inhibition also contain phenol, may have effects COX-1 and COX-2 (9). Overall, acetaminophen appears to act only at site-specific in vivo sites. In the gastric mucosa, acetaminophen poorly inhibits COX but in blood, platelets are at least partially inhibited by acetaminophen (9). The majority of studies promoting the existence of a "COX-3" are rooted in an effort to explain the pharmacology of acetaminophen. It is a general belief that another COX enzyme is selectively inhibited by acetaminophen and thus this enzyme plays a role in central prostaglandin production, which in turn produces pain and fever. The inhibition of a distinct COX enzyme by acetaminophen as first postulated in 1972 (4), could represent a primary central mechanism by which acetaminophen and possibly other analgesics decrease pain and fever.
COX-SPLICE VARIANTS
It is well established that alternative splicing of mRNA is functionally significant. In addition to being one of the first groups (3, 10) responsible for elucidating the COX-1 and COX-2 isoforms, Xie et al. (10) also importantly described a splice variant of COX-2 in chicken cells with retention of a 550 (base pair) bp intron between exons 1 and 2. Another splice variant of COX-1 in human fibroblasts was subsequently reported by Diaz (11).
In 1993, Dewitt and Meade (12) made an interesting observation that serum treatment of 3T3 cells induces COX-1 mRNA levels. However, no concomitant increase in COX-1 protein was detected. Similar findings in rat tracheal epithelial (RTE) cells were found whereby 12-myristate 13-acetate diester treatment induced COX-1 mRNA. However, Western blot analysis could not unequivocally demonstrate protein induction in these cells, despite an increase in COX-1 mRNA being detected (13). Interestingly, RTE cells also show an aberrantly spliced COX form that lacks codon 1-36. This COX-1 mRNA induction without evidence of protein production could be due to specific induction of splice variant COX-1 V1 mRNA that does not produce protein, or that the variant protein does not recognize the non-specific antibody used to detect the protein.
In EGC-6, RTE cells, and rat fibroblasts cells, the retention of an intron and a COX-1 splice variant was described (13). In other investigations, a COX-1 transcript of similar length 5.2 kilobases (kb) was previously shown to be present in endothelial cells (14). Plant and Laneuville (15) also describe three transcripts of COX-1 genes (2.8, 4.5 and 5.1kb) as well as two transcripts of the COX-2 gene (2.8 and 4.5 kb). All of these studies uniformly suggest the existence of COX-isoform splice variants with a tissue specific profile of expression.
These initial studies demonstrating COX splice variants consistently utilized splice variant nomenclature while referring to these new COX variants as they were discovered. In 1999, a COX variant COX-2b was discovered although it was suggested it might be considered a third COX (5). Subsequently, the existence of an additional COX isozyme was suggested by others. (16-17) Willoughby et al. postulated that an inducible isoenzyme expressed in later, resolving stages of inflammation was "COX-3" (16). These papers suggest that "COX-3" is a variant of COX-2 that includes the COX-2-specific carboxy terminus; however, each of these publications utilized the designation "COX-3".
A newly discovered COX variant enzyme in 2002 (6) was also recently dubbed "COX-3", even though it derives from the same gene as COX-1, because it was suggested that experience has shown that the important differences between COX-1 and COX-2 are pharmacological rather than genetic (6). Subsequent articles, commentaries, letters, and editorials have referred to this variant as a "COX-3" (18-26). The use of the term "COX-3" is confusing, as it has been applied to at least two different COX variants. We believe that the moniker "COX-3" should be reserved for the product of an independent third COX gene which clearly has not been yet identified in any species. Furthermore, differences between COX-1 and COX-2 are both genetic and pharmacological. It is, therefore, necessary for the scientific community to develop suitable nomenclature for future COX advancements based on genetic isoforms rather than simply function as physiological and pathophysiological functions of COX appear to have extensive overlaps. Moreover, as the expression and sequence of this most recently described COX variant (6) closely resembles COX-1, the nomenclature should also reflect this.[Figure 1] In this article the variant COX enzyme described by Chandrasekharan et al. in 2002 (6) will be termed COX-1V1 in accordance with the accepted naming pattern utilized in the literature for COX variants between 1991-1999.
COX-2 SPLICE VARIANT 1 (COX-2V1)
In 1999, Simmons et al. (6) suggested that a third, acetaminophen-sensitive isozyme of COX could be induced in a murine macrophage cell line (J774.2) subject to chronic COX inhibition with indomethacin, aspirin, racemic flurbiprofen or diclofenac in a concentration dependent manner. Diclofenac appeared to induce a COX-2 variant protein, furthermore, lipolysaccharide also induced a COX-2 protein, however, only the diclofenac-induced COX-2 variant was inhibited by acetaminophen. This new enzyme was not isolated or cloned, but it was determined to be immunologically related to COX-2. Most COX-2 antibodies are designed to recognize some part of the amino acid sequence between residues 570-598, which is unique to COX-2. There is an absence of structural information provided for the remainder of the COX sequence in this manuscript, except for some pharmacology studies showing differences in NSAID binding and inhibition. This COX variant was less sensitive to inhibition by racemic flurbiprofen or tolfenamic acid both of which are potent inhibitors of COX-1 and COX-2. Interestingly, aspirin did not inhibit this diclofenac-induced variant enzyme, suggesting that the active site of this variant enzyme may be altered. It also remains possible that a similarly induced enzyme in vivo is a target for acetaminophen, which leads to subsequent analgesic, and anti-pyretic activity for this drug. The changes that are responsible for resulting in this COX variant are not known. This enzyme was speculated to be a COX-2 variant or a third COX. There does not seem to have been any published follow up investigations concerning this COX-2 variant in the literature.
COX-1 SPLICE VARIANT 1 (COX-1V1 )
In 2002, acetaminophen was shown to inhibit another COX strongly expressed in the brain and it was suggested that it might be involved in mediating pain and perhaps fever (6). This COX-1 variant was discovered in dogs when two forms of mRNA for COX-1 were found in dog brain. A sequence of the proposed "COX-3" was published and attributed to a clone of the COX-1 gene with alternative splicing, resulting in the retention of intron-1 in the mature mRNA. Translation of the RNA results in a COX isozyme with an N-terminal extension due to intron-1 and the retained signal peptide. This COX-1 variant (COX-1V1) named "COX-3" in this publication was then expressed in baculovirus insect cells to see if this different mRNA would code for a functional protein. The insect cells produced a novel COX that produced prostaglandins. The inhibition parameters of the recombinant protein to various analgesics and anti-inflammatory drugs were reported. In addition, two smaller forms of COX-1, "partial COX", were also determined (PCOX-1a and PCOX-1b). Two concentrations of arachidonic acid were used (5 m M and 30 m M) in order to produce PGE2 . At the lower concentration acetaminophen was only a marginally more potent inhibitor of prostaglandin synthesis in the insect cells containing COX-1V1 than in cells containing mouse COX-1 but was much more important in cells containing mouse COX-2. At high concentrations of arachidonic acid COX-1V1, inhibition potency was reduced and eliminated for COX-1 and COX-2. Further, investigations at low concentrations of arachidonic acid or following stimulation with other stimulants are required to characterize the selectivity of acetaminophen and other selective inhibitors of this COX variant isozyme.
It is suggested that the COX-1 gene mRNA is translated to COX-1 protein in dogs and humans (6). There is alternative splicing in the COX-1 gene, so some cells remove a stretch of the mRNA sequence intron 1. Other cells included intron 1 to form a COX-1V1 protein. Therefore, COX-1V1, dubbed "COX-3", has the same sequence as COX-1 except that it retains intron 1. This retention of intron 1 inserts 30-34 amino acids depending on the mammalian species. The retention of intron 1 could possibly change protein folding and active site conformation. PCOX-1a is made from the COX-1 gene and retains intron 1; however, exons 5-8 are deleted. PCOX-1b differs in size by 90 nucleotides to PCOX-1a as it also lacks intron 1. Both PCOX-1a and -1b mRNA was expressed in dog brain cortex (6).
Subsequently, the presence of COX-1V1 was examined in various human tissues for the presence of COX-1V1 mRNA. In humans, COX-1V1 mRNA expression has shown to be tissue specific and most abundant in the heart and cerebral cortex at ~5% of normal COX-1; another faint mRNA band was also detected. COX-1V1 appears to be more sensitive than COX-1 or COX-2 to inhibition by acetaminophen and is much more sensitive to diclofenac, indomethacin, racemic ibuprofen and aspirin.
However, production of PGE2 from exogenous arachidonic acid, which is indicative of basal COX activities of the three Sf9 constructs, was significantly different. Furthermore, two different species including dog and mice COX enzymes were employed. The COX-1V1 construct was approximately 20% as active as the COX-1 construct and was only 4% as active as the COX-2 construct. Therefore, it is difficult to know whether acetaminophen truly has a greater ability to inhibit COX-1V1>COX-1>>COX-2, or if the 50% inhibitory concentrations (IC 50 's) merely reflect the variable activities of the Sf9 constructs. Future studies using constructs with equivalent arachidonic acid utilization should be employed. Employing purified (or microsomal) preparations of the different COX isoforms exhibiting similar specific activities would also allow a better comparison of the inhibitory potency of acetaminophen and NSAIDs against the isoforms. Furthermore, it should be noted that in the initial report canine (COX-1V1) was compared to murine (COX-1) and murine COX-2 expressed by transected cells. Interestingly, the DNA sequencing of intron 1 from dog, human and mouse COX-1 genes displays a high degree of conservation.
Predictably, these interesting findings were followed by various commentaries, reports, and editorials (18-27). Each commentary also used the "COX-3" appellative; however, some commentaries suggest this new COX variant is best described as a COX-1b.
COX-1V1 : FURTHER INVESTIGATIONS
O'Brien's group in Australia (28) followed up on the COX splice variant work of Kitzler (13) in rats and investigated the effect of ageing on COX gene expression in the normal rat stomach. COX-1V1 mRNA levels were lower than COX-1 mRNA levels. COX-1V1 mRNA levels were elevated in adult rats but not induced after acute gastric injury (28). In a rat colorectal cancer model COX-1V1 mRNA levels were elevated in colorectal tumors and reduced after the NSAIDs (sulindac and sulindac sulphone) and COX-2 inhibitor (celecoxib) treatment to levels observed in normal colonic mucosa (29). These data suggests that COX-1V1 may be regulated by NSAID treatment and may be affected by the ageing process (13, 29).
The scientific literature is now reacting to the findings exploring the expression patterns of COX variants. The expression of putative rat (COX-1V1) mRNA in primary cultures of neurons, astrocytes, endothelial cells, pericytes, and choroidal endothelial cells was demonstrated in rat brain. mRNA of COX-1V1 was expressed in all cells except neurons with cerebral endothelial cells having the highest expression. This expression did not change after lipopolysachharide challenge suggesting this enzyme may be constitutive (30). COX-1V1 mRNA was ~13% the levels of COX-1 mRNA. A follow-up study by the same group in rat central nervous system demonstrated COX-1V1 expression was highest in the choroid plexus and spinal cord followed by pituitary gland, hypothalamus, hippocampus, medulla, cerebellum and cortex. The mRNA levels were higher in major brain arteries and dramatically higher in brain microvessels and appeared to relate to vascular density of a given brain region. COX-1V1 mRNA was ~30% of the levels of COX-1 mRNA in brain microvessels (31). It remains unknown if this COX-1V1 mRNA encodes a functional or truncated protein in mice or rats. These initial findings do not rule out the possibility that COX-1V1 can be induced by other stimuli or under other pathophysiological conditions.
A recent publication suggests COX-1V1 mRNA is present in heart, aorta, lung, brain and cerebellum but not in whole blood of rat (33). The Western blotting using a commercially available antibody raised against canine (COX-1V1 ) did not detect any immunoreactive proteins in rat. Acetaminophen was not a selective inhibitor of "COX" activities in the central nervous system of the rat. This data is consistent with the apparent impossibility for expression of COX active protein from COX-1V1 mRNA in the rat due to frame shift error. Comparing acetaminophen to other NSAIDs provided no evidence for acetaminophen inhibitable COX activity in cerebellum or brain in the rat (32).
The expression of mRNA encoding a COX-1V1 in the mouse central nervous system has been suggested (33). Tissues from C57BL/6 mice were prepared from cortical tissue after 8 hours intra cortical interleukin (IL)-1b or vehicle injections and from spleen homogenates, primary mouse astrocyte and microglial cultures. All tissues exhibited COX-1V1 mRNA but at lower relative amounts than COX-1 mRNA. The COX-1V1 mRNA was not significantly induced by an acute inflammatory stimulus IL-1b. In the mouse, the COX-1V1 product revealed that the nucleotides of the COX-1 intron are present and are retained in frame.
"COX-3": A THEORY WITHOUT EXPERIMENTAL EVIDENCE?
Some investigators are now referring to acetaminophen as a "COX-3" inhibitor or even a "selective COX-3 inhibitor" in rat studies (34-35). The use of this term has been undertaken without any proof of a functional "COX-3" rat protein or that acetaminophen functionally works selectively through such an enzyme in vivo in any species.
Willoughby et al. (16) suggested a third physiological form of COX that may be a separate form of COX-1 and -2 based on a late-induced enzyme appearing 48 hours after the start of the inflammatory process, which may produce prostaglandins involved in the resolution of inflammation. Early produced COX-2 expresses PGE2 while late appearing COX-2 synthesizes mainly PGD2 (36). It is hypothesized that COX-2 functions in resolution of acute inflammatory responses and that "COX-3" is turned on later in inflammation and may be involved in the biosynthesis of endogenous anti-inflammatory mediators. It is speculated that such an enzyme may induce cyclopenetanone prostaglandins and resolvins, which can be triggered by aspirin treatment (37-38).
COX SINGLE NUCLEOTIDE POLYMORPHISMS
Single nucleotide polymorphisms (SNPs) may be useful in identifying candidate disease genes and individuals at risk of disease. Polymorphisms in the COX-1 or -2 genes could alter enzyme expression, function, and response to anti-inflammatory drugs. In addition, increased risks for cancer, hypertension, other diseases and occurrence of greater side effects due to drugs are also possible with different SNP's. Both the COX-1 and COX-2 gene have demonstrated SNP sequences with rare polymorphisms being identified (39-40). The demonstration of the low numbers of and rare allelic variation in functionally important polymorphisms of COX-1 and COX-2 genes may suggest that because of the critical roles of these enzymes, SNPs are not likely to develop.
For the COX-1 isoform there were at least 18 SNPs identified with seven resulting in amino-acid changes. Preliminary allele frequencies suggest these SNPs were all rare and found in less than 4% of the population (41). Interestingly, two SNPs heterozygotes (A-842G and C50T) demonstrated significantly greater inhibition of GH2 inhibition by aspirin compared to common allele homozygotes. However, any pharmacological or clinical relevance of this SNP has not been established (40).
For the COX-2 isoform, over twenty different SNPs have been detected (42, 43). Preliminary studies of some of these variants did not reveal any pharmacological distinctiveness and their allele frequency estimates suggest that they are very uncommon. In addition, none of these allelic variants segregated with a particular colorectal disease phenotype (42, 43). However, more recently, several polymorphisms in the coding region for COX-1 were examined and an increased risk for individuals heterozygous for the COX-1 SNP L15L16-Ldel allele and adenoma risk among non-users of NSAIDs was identified. Moreover, a reduced risk of adenoma among users of NSAIDs for P17L was reported.(44)
More information on COX-1 and COX-2 SNP's can be located at the National Centre for Biotechnology SNP Database (http://www.ncbi.nlm.nih.gov/SNP).
COX-1 SPLICE VARIANT ANTIBODIES AND PRIMERS
It is important that pharmaceutical companies develop COX reagents, antibodies, primers and screening kits specific for each variant COX found. Several companies now sell COX-1V1 antibodies for mouse, human, and dog (Table 1).
Table 1: Antibodies to COX-1V1
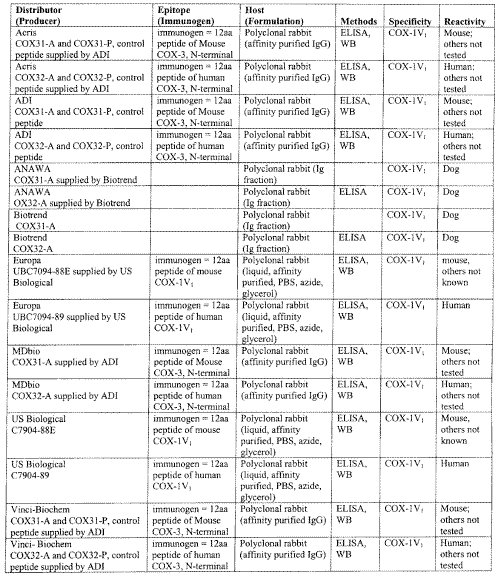
These antibodies may only be used in a species-selective manner, as those raised against one species of COX-1V1 may possibly not react with other species COX-1V1 proteins.
In addition, there are published functional primers for rat, mouse, dog and human studies available (Tables 1 and 2).
Table 2: PCR Primers Specific for COX-1V1 in Various Species.
Specific inhibitors of COX-1V1 are currently being developed by several companies, and provision of them to basic researchers as scientific tools may assist in determining the functional roles of the COX variants.
DISCUSSION
Pre-clinical animal models have dramatically increased our understanding of the functional roles of COX-1 and COX-2 inhibition. Animals are often used as models for human pain, inflammation, toxicity, and cancer. It is important to understand the expression and functional roles of COX-variants in animals in order to develop and test new compounds in the most appropriate animal model for the development of new drugs.
Recent work using transgenic knockout mice deficient in the isozymes for COX have shed some light on the specific signalling roles of the two prostaglandin biosynthetic pathways defined by these enzymes. The functional roles of prostaglandins derived from COX-1, COX-2, and their variants need to be further elucidated. It is entirely possible that some of the effects of NSAIDs will be related to their ability to suppress COX-1V1 and that the prostanoids derived via COX-1V1 contribute to inflammation, pain, fever, Anti-Alzheimer's and anti-cancer effects. Furthermore, it is possible that PCOXs will also have other relevant physiological and pathophysiological functions besides COX. Currently, COX-1 knockout mice are deficient in both COX-1 and COX-1V1. No specific knockout mice for each COX variant have been developed. To functionally test the hypothesis that (COX-1V1 ) is an acetaminophen-inhibitable COX is to determine the effect of this drug in COX-1 knockout mice. The development of specific COX-1V1 knockout mice may be technically challenging and may never eventuate.
Many tumours have been shown to over-express COX-2. Specific COX-2 inhibitors are currently being explored for treatment efficacy in various cancers. Celecoxib has received Food and Drug Administration approval for the treatment of familial adenomatous polyposis. Interestingly, there are some other cancers that over express COX-1 including ovarian adenocarcinomas and cervical carcinomas (45, 46). COX-1 activity also potentiates the differentiation of human acute promyelocytic leukemia (47). Whether the COX-1V11 variants is expressed in some cancer and its specific inhibition will demonstrate beneficial anti-cancer activity remains to be determined.
In mice lacking the COX-1 gene, pyresis is not blunted. Neither the COX-1 gene nor the COX-1 protein appears to underlie pyresis. COX-2 knockout mice have blunted pyretic responses and COX-2 inhibitors are effective anti-pyretic agents (48, 49). In addition, the anatomical site of fever regulation is the hypothalamus and COX-1V 1 is expressed in the cerebral cortex. However, it remains possible that COX-1V11 could contribute to fever response and underlie pyresis.
It is now well established that COX-2 inhibitors have clinically demonstrated the ability to reduce inflammation and analgesic response in clinical use. The role of COX-1 has been conceptually neglected in inflammation (50, 51). A role for COX-1 in fever may exist and a role of COX-1 in pain may exist. In a model of postoperative pain, COX-1 immunoreactivity increased in glial cells of the spinal cord and intrathecal SC-560 (a specific COX-1 inhibitor) was effective in inhibiting pain (52). Whether targeting COX-1V1 specifically in the spinal cord or development of a specific COX-1V 1 inhibitor could prove an effective strategy for certain types of analgesia and inflammation remains possible.
Interestingly, we have previously demonstrated that compensatory increases in COX enzymes can occur. For instance, COX-2 may be up regulated when COX-1 is inhibited in the stomach (53). How, when and why COX-1V1 is increased or decreased or co-regulated with other COX's and the relevance of these compensatory increases to physiology and pathophysiology remains to be elucidated.
Dinchuck et al. (54) cloned various COX-1 cDNA's and demonstrated that some clones did indeed retain intron 1. However, they also found that their clones agreed with human COX-1 intron 1 sequence available from human genome sequencing effort (accession NT_017568). Intron 1 is 94 nucleotides long and shifts the remaining COX-1 variant out of frame with respect to open reading (51, 54). In the rat retention of intron 1 in rat COX-1 would also shift the COX-1V1 sequence out of frame with respect to the open reading frame resulting in completely different proteins (32). However, Simmons 2003 (55) suggests alternative polydentation occurs in human COX-1V1, and in spite of the fact that intron 1 is out of frame; at least part of the intron is retained in COX-1V1 proteins. It is apparent that intron-1 is out of frame in rodents and some alternative processing event overcomes the effect of the frame shift in intron-1 to produce intron-1 antibody-recognized proteins (54). COX-1 polymorphisms were recently screened for the human COX-1 gene including the intron 1 region and no polymorphisms or deletions in intron 1 were determined (41).
Compensation by ribosomal frame shifting is possible, although a questionable explanation in producing a functional COX-1V1 protein. A 65 kda protein in human aorta postulate to be COX-1V1 and a ~53 and 50 kD protein postulated to be PCOX1a and b at ~25%, the level of COX-1 has been determined.(6) Interestingly, COX-1V1 is smaller than predicted for a glycosylated protein to the same extent as COX-1 which could suggest hypo-glycosylation or other differences. Currently, the activities of PCOX1-a & b are not known (6, 55).
It is apparent that more investigations must be undertaken on COX-1V1 as some of the published sequences differ by one nucleotide in intron 1 and hence are out of frame. Whether there are indeed genuine polymorphisms or sequencing errors in all relevant species utilized in drug development should be ascertained. Whether ribosomal frame shift occurs in all species and is responsible for the production of a functional COX-1V1 protein needs to be further detailed.
Importantly with the discovery of new COX variants, it must be recognized that previous voluminous data on COX-2/COX-1 ratio inhibition in fact involves COX-2 and its variants:COX-1 and its variants. COX inhibition data with NSAIDs and COX-2 inhibitors must bear in mind the discovery of new COX variants, as previous literature data do not reflect specificity to each currently known isoform and variant. For instance the presence of a splice variant may be experimentally important in that it could lead to the overestimation of the amount of COX-1 or COX-2 mRNA on polymerase chain reaction (PCR) with primers that do not distinguish the splice variant from COX-1 or COX-2 or in Northern blots probed with full-length COX-1cDNA. This could lead to underestimation of COX-1 or -2 mRNA and an overestimation of induction of functional COX mRNA on the Northern blots. It is possible that investigations that show no significant induction of COX-1 or COX-2 mRNA will be because high basal levels of COX splice variant mRNA mask induction of COX-1 or -2 mRNA.
CONCLUSIONS
Some recent and exciting scientific discoveries surrounding COX have led to a further conceptual understanding and much confusion in the scientific literature about formation, action, and nomenclature of these prostanoid-producing enzymes (Figure 1). Currently, there remain just a handful of research papers available regarding these COX variant discoveries. Further, two of these well-recognized and publicised research papers (5, 6) point scientists in two very different directional pathways. Temporally "COX-3" was first demonstrated to be a COX-2 variant (5) then subsequently a COX-1 variant. Ultimately, however, there is not any concrete scientific evidence for an actual third independent COX gene in the literature to date. Although COX isoenzymes are currently derived from two distinct genes, this does not rule out the possibility of other undiscovered COX genes. With the wisdom of retrospection we can be confident that both COX-1, COX-2, and their variants will all demonstrate physiological and pathological roles. Interestingly, there is organ and tissue-specific alternative splicing of COX-1V1, which may prove to be an important method of modulating COX-1 activity. Whether or not a consequential target of acetaminophen activity has been actually discovered still remains to be seen.
Perhaps the most pertinent scientific question that remains to be comprehended surrounding COX is why are there so many. The determination of this perplexity and a swift riposte may lie in our eventual understanding of the natural substrates for COX other than arachidonic acid. Optimistically, these COX variants may eventually lead to more effective pain relievers, anti-inflammatories, anti-pyretics, anti-cancer agents, or treatments for Alzheimer's and other conditions. Undoubtedly, all of these potential indications and avenues will surely be exploited (if possible) by pharmaceutical companies in the future.
References
Vane, J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Bio, 231(25):232-235, 1971.
Fu, J.Y., Masferrer, J.L., Seibert, K., Raz, A., Needleman, P. The induction and suppression of prostaglandin H2 synthase (cylooxygenase) in human monocytes. J Biol Chem, 265(28):1727-1740, 1990.
Simon LS. COX-2 inhibitors. Are they nonsteroidal anti-inflammatory drugs with a better safety profile?Gastroenterol Clin North Am. 30:1011-1025, 2001.
Flower, R.J., Vane, J.R. Inhibition of prostaglandin synthetase in brain explains the anti-pyretic activity of paracetamol (4-acetamidophenol). Nature, 240(5381):410-13, 1972.
Simmons, D.L., Botting, R.M., Robertson, M., Madsen, M.L., Vane, J.R. Induction of an acetaminophen-sensitive cyclooxygenase with reduced sensitivity to nonsteroid antiinflammatory drugs. Proc Natl Acad Sci U S A, 96(6):3275-80,1999.
Chandrasekharan, N.V., Dai, H., Roos, K.L., Evanson, N.K., Tomsik, J., Elton, T.S., Simmons, D.L. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A, 99(21):13926-31, 2002.
Ouellet, M., Percival, M.D. Mechanism of acetaminophen inhibition of cyclooxygenase isoforms. Arch Biochem Biophys, 387(2):273-80, 2001.
Prescott, L.F., Paracetamol: past, present and future. Am J Ther, 7:143-147, 2000.
Graham, G.G., and Scott, K. F., Mechanisms of action of paracetamol and related analgesics. Inflammopharmacology, 11(4-6): 401-413, 2003.
Xie, W.L., Chipman, J.G., Robertson, D.L., Erikson, R.L., Simmons, D.L. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci U S A, 88:2692-2696, 1991.
Diaz, A., Reginato, A.M., Jimenez, S.A. Alternative splicing of human prostaglandin G/H synthase mRNA and evidence of differential regulation of the resulting transcripts by transforming growth factor beta 1, interleukin 1 beta, and tumor necrosis factor alpha. J Biol Chem, 267(15):10816-22, 1992.
DeWitt, D.L., Meade, E.A. Serum and glucocorticoid regulation of gene transcription and expression of the prostaglandin H synthase-1 and prostaglandin H synthase-2 isozymes. Arch Biochem Biophys, 306(1):94-102, 1993.
Kitzler, J., Hill, E., Hardman, R., Reddy, N., Philpot, R., Eling, T.E. Analysis and quantitation of splicing variants of the TPA-inducible PGHS-1 mRNA in rat tracheal epithelial cells. Arch Biochem Biophys, 316(2):856-63, 1995.
Plant, M.H., Laneuville, O. Characterization of a novel transcript of prostaglandin endoperoxide H synthase 1 with a tissue-specific profile of expression. Biochem J, 344:677-685, 1999.
Hla, T. Molecular characterization of the 5.2 KB isoform of the human cyclooxygenase-1 transcript. Prostaglandins, 51(1):81-5. 1996.
Willoughby, D.A., Moore, A.R., Colville-Nash P.R. COX-1, COX-2 and COX-3 and the future treatment of chronic inflammatory disease. Lancet, 355(9204):646-648, 2000.
Botting, R.M. Mechanism of action of acetaminophen: is there a cyclooxygenase 3? Clin Infect Dis, 31 Suppl 5:S202-10, 2000.
Wickelgren, I. Enzyme might relieve headache. Science, 297(5589):1976, 2002
Bazan, N.G., Flower, R.J. Medicine: lipid signals in pain control. Nature, 420(6912):135-8, 2002.
Schwab, J.M., Schluesener H.J., Meyermann, R., Serhan, C.N. COX-3 the enzyme and the concept: steps towards highly specialized pathways and precision therapeutics? Prostaglandins Leukot Essent Fatty Acids, 69(5):339-43, 2003.
Schwab, J.M., Schluesener, H.J., Laufer, S. COX-3: just another COX or the solitary elusive target of paracetamol? Lancet, 361(9362):981-2, 2003.
Smith, A. Time for a new COX. Nature Reviews, 1:839, 2002.
Warner, T.D., Mitchell, J.A. Cyclooxygenase-3 (COX-3): filling in the gaps toward a COX continuum? Proc Natl Acad Sci U S A, 99(21):13371-3, 2002.
Warner, T.D., Mitchell, J.A. Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. FASEB J, 18:790-804, 2004.
van der Donk, W.A., Tsai, A.L., Kulmacz, R.J. The cyclooxygenase reaction mechanism. Biochemistry, 41:15451-15458, 2002.
Botting, R. COX-1 and COX-3 inhibitors. Thrombosis Research, 110:269-272, 2003.
Senior, K., Homing in on COX-3 – the elusive target of paracetamol. The Lancet, 1:399, November 2002.
Vogiagis, D., Glare, E.M., Misajon, A., Brown, W., O'Brien, P.E. Cyclooxygenase-1 and an alternatively spliced mRNA in the rat stomach: effects of aging and ulcers. Am J Physiol Gastrointest Liver Physiol, 278(5):G820-7, 2000.
Vogiagis, D., Brown, W., Glare, E.M., O'Brien, P.E. Rat colorectal tumours treated with a range of non-steroidal anti-inflammatory drugs show altered cyclooxygenase-2 and cyclooxygenase-1 splice variant mRNA expression levels. Carcinogenesis, 22(6):869-74, 2001.
Kis, B., Snipes, J.A., Isse, T., Nagy, K., Busija, D.W. Putative cyclooxygenase-3 expression in rat brain cells. J Cereb Blood Flow Metab, 23(11):1287-92, 2003.
Kis, B., Snipes, A., Bari, F., Busija, D.W. Regional distribution of cyclooxygenase-3 mRNA in the rat central nervous system. Brain Res Mol Brain Res, 126(1):78-80, 2004.
Warner, T.D., Vojnovic, I., Giuliano, F., Jiminez, R., Bishop-Bailey, D., Mitchell, J.A. Cyclooxygenases-1, 2 and 3 and the production of prostaglandin I2: investigating the activities of acetaminophen and cyclooxygenase-2-selective inhibitors in rat tissues. J Pharmacol Exp Ther, In Press, May 17 2004 [Epub ahead of print] PMID: 15148345 [PubMed - as supplied by publisher].
Shaftel, S.S., Olschowka, J.A., Hurley, S.D., Moore,A.H., O'Banion, M.K. COX-3: a splice variant of cyclooxygenase-1 in mouse neural tissue and cells. Brain Res Mol Brain Res, 119(2):213-5, Nov 26 2003.
Gryglewski, R.J., Swies, J., Uracz, W., Chlopicki, S., Marcinkiewicz, E. Mechanisms of angiotensin-converting enzyme inhibitor induced thrombolysis in Wistar rats. Thromb Res, 110(5-6):323-9, 2003.
Laudanno, O.M., San Miguel, P., Aramberry, L.J., Cesolari, J.A. Mechanism of inhibition Of COX-2 and COX-3 in gastrointestinal damage induced by NSAID in rats. Acta Gastroenterol Latinoam, 33(4):183-5, 2003.
Gilroy, D.W. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med, 5(6):698-701, 1999.
Serhan, C.N., Hong, S., Gronert, K., Colgan, S.P., Devchand, P.R., Mirick, G., Moussignac, R.L. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med, 196(8):1025-37, 2002.
Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxin biosynthesis: an update and role in anti-inflammation and pro-resolution. Prostaglandins Other Lipid Mediat., 68-69:433-55, 2002.
Fritsche, E., Baek, S.J., King, L.M., Zeldin, D.C., Eling, T.E., Bell, D.A. Functional characterization of cyclooxygenase-2 polymorphisms. J Pharmacol Exp Ther, 299(2):468-76, 2001.
Halushka, M.K., Walker, L.P., Halushka, P.V. Genetic variation in cyclooxygenase 1: effects on response to aspirin. Clin Pharmacol Ther, 73(1):122-30, Jan 2003.
Ulrich, C.M., Bigler, J., Sibert, J., Greene, E.A., Sparks, R., Carlson, C.S., Potter, J.D. Cyclooxygenase 1 (COX1) polymorphisms in African-American and Caucasian populations. Hum Mutat, 20(5):409-10, 2002.
Humar, B., Giovanoli, O., Wolf, A., Attenhofer, M., Bendik, I., Meier, R., Muller, H., Dobbie, Z. Germline alterations in the cyclooxygenase-2 gene are not associated with the development of extracolonic manifestations in a large swiss familial adenomatous polyposis kindred. Int J Cancer, 87(6):812-7, 2000.
Spirio, L.N., Dixon, D.A., Robertson, J., Robertson, M., Barrows, J., Traer, E., Burt, R.W., Leppert, M.F., White, R., Prescott, S.M. The inducible prostaglandin biosynthetic enzyme, cyclooxygenase 2, is not mutated in patients with attenuated adenomatous polyposis coli. Cancer Res, 58(21):4909-12, 1998.
Ulrich, C.M., Bigler, J., Sparks, R., Whitton, J., Sibert, J.G., Goode, E.L., Yasui, Y., Potter, J.D. Polymorphisms in PTGS1 (=COX-1) and risk of colorectal polyps. Cancer Epidemiol Biomarkers Prev, 13(5):889-93, 2004.
Dore, M., Cote, L.C., Mitchell, A., Sirois, J. Expression of prostaglandin G/H synthase type 1, but not type 2, in human ovarian adenocarcinomas. J Histochem Cytochem, 46(1):77-84, 1998.
Sales, K.J., Katz, A.A., Howard, B., Soeters, R.P., Millar, R.P., Jabbour, H.N., Soeters, R.P. Cyclooxygenase-1 is up-regulated in cervical carcinomas: autocrine/paracrine regulation of cyclooxygenase-2, prostaglandin e receptors, and angiogenic factors by cyclooxygenase-1. Cancer Res, 62(2):424-32, 2002.
Rocca, B., Morosetti, R., Habib, A., Maggiano, N., Zassadowski, F., Ciabattoni, G., Chomienne, C., Papp, B., Ranelletti, F.O. Cyclooxygenase-1, but not –2, is upregulated in NB4 leukemic cells and human primary promyelocytic blasts during differentiation. Leukemia, 1-4, 2004.
Li, S., Wang, Y., Matsumura, K., Ballou, L.R., Morham, S.G., Blatteis, C.M. The febrile response to lipopolysaccharide is blocked in cyclooxygenase-2(-/-), but not in cyclooxygenase-1(-/-) mice. Brain Res, 825(1-2):86-94, 1999.
Schwartz, J.I., Chan, C.C., Mukhopadhyay, S., McBride, K.J., Jones, T.M., Adcock, S., Moritz, C., Hedges, J., Grasing, K., Dobratz, D., Cohen, R.A., Davidson, M.H., Bachmann, K.A., Gertz, B.J. Cyclooxygenase-2 inhibition by rofecoxib reverses naturally occurring fever in humans. Clin Pharmacol Ther, 65(6):653-60, 1999.
Schwab, J.M., Schluesener, H.J. Cyclooygenases and central nervous system inflammation: concenptual neglect of cylooxygenase 1. Arch Neurol, 60(4):630-632, 2003.
Schwab, J.M., Beiter, T., Linder, J.U., Laufer, S., Schulz, J.E., Meyermann, R., Schluesener, H.J. COX-3--a virtual pain target in humans? FASEB J, 17(15):2174-5, 2003.
Zhu, X., Conklin, D., Eisenach, J.C. Cyclooxygenase-1 in the spinal cord plays an important role in postoperative pain. Pain, 104(1-2):15-23, 2003.
Davies, N.M., Sharkey, K.A., Asfaha, S., Macnaughton, W.K., Wallace, J.L. Aspirin causes rapid up-regulation of cyclo-oxygenase-2 expression in the stomach of rats. Aliment Pharmacol Ther, 11(6):1101-8, 1997.
Dinchuck, J.E., Liu, R.Q., Trzaskos, J.M. COX-3 in the wrong frame in mind. Immunology Letter, 86(9):121, 2003.
Simmons, D.L. Variants of cylooxygenase-1 and their roles in medicine. Thrombosis Research, 110:265-268, 2003.
Corresponding Author: Neal M. Davies, Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, PO Box 646534, Pullman, WA 99164-6534, USA. ndavies@wsu.edu
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps

