J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 7(1):47-54, 2004
Hepatic disposition of cyclosporine A in isolated perfused rat livers.
Reza Mehvar1, Anjaneya P. Chimalakonda
School of Pharmacy, Texas Tech University Health Sciences Center, Amarillo, Texas, USAReceived 03 February 2004, Revised 10 February 2004, Accepted 10 February 2004, Published 17 February 2004
PDF Version
Abstract
PURPOSE: To develop an isolated perfused rat liver model to study the hepatic disposition of cyclosporine A (CyA) in both sexes. METHODS. Livers were isolated from male (n = 6) and female (n = 7) rats and perfused with a physiological buffer in a single-pass manner. A bolus 1-mg dose of CyA was injected into the inlet catheter and periodical samples (0-15 min) were collected from the outlet perfusate. The concentrations of CyA in the outlet perfusate, collected bile (0-15 min), and liver tissue (at the end of perfusion) were quantitated by HPLC and subjected to statistical moment analysis. RESULTS. The dilution curves of CyA in the outlet perfusate exhibited unusually long terminal phases due to large volume of distribution of the drug (~100 mL/g) and its slow release from binding sites in the liver (net release rate constant of ~0.020 min-1 ). This was in contrast to the rapid uptake of the drug, indicated by significant amounts of the intact drug (>40%) taken up during one single pass through the liver. Consequently, the liver tissue:perfusate distribution ratio of CyA was very high (~220). No significant differences were found between the male and female livers in any of the estimated parameters. CONCLUSIONS. The tissue binding of cyclosporine A is substantial, slowly reversible, and gender-independent in isolated perfused rat livers.
Introduction
Cyclosporine A (CyA) is a cyclic peptide with a potent immunosuppressive activity, which has been partly responsible for the rapid advancement of organ transplantation during recent years. The drug is extensively metabolized in the liver (1, 2), and the metabolites are mostly excreted into the bile (3). Additionally, CyA may be metabolized by enzymes in the gastrointestinal tract (4-6). Pharmacokinetics of CyA have been studied extensively in both humans (3, 7) and animals (8-12) and are characterized by a low clearance and a high volume of distribution. Despite low hepatic extraction ratio, the systemically-administered drug accumulates substantially in the liver of rats (8, 10, 11), where it binds to proteins, including its effect target protein cyclophilin (13); the binding of CyA to cyclophilin is responsible for the blockade of the activity of calcineurin which leads to immunosuppression (14). Therefore, delineation of the hepatic disposition of CyA is crucial for understanding the pharmacokinetics and dynamics of the drug.
Isolated perfused rat liver (IPRL) is a model that has been used extensively for the investigations of the hepatic disposition of drugs. The model is devoid of in vivo factors that may confound the drug hepatic disposition while preserving the integrity of the intact organ. Despite significant binding and/or accumulation of CyA in the liver after in vivo administration (8, 10, 11) and in the hepatocytes after in vitro incubation (13), the kinetics of binding, including binding reversibility, of CyA in the intact liver are not known. A recirculating IPRL model, which was previously used (15) to study the effects of LDL and ethinyl estradiol on the metabolism of CyA, showed irregular CyA profiles in the outlet perfusate, making it difficult for use in binding studies. Therefore, the aim of this study was to investigate the suitability of a single-pass IPRL model and statistical moment theory (16) for delineation of the hepatic disposition of CyA. Because there are reports suggesting gender differences in the in vivo hepatic disposition of CyA in rats (10), the studies were conducted in both male and female livers.
Materials and Methods
Chemicals
Cyclosporine (Cyclosporine Injection, USP, 50 mg/mL) was obtained from Bedford Laboratories (Bedford, OH, USA). Tamoxifen, fluorescein-labeled dextran 70 kD (FD-70), and kits for measurement of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were obtained from Sigma Chemical Co. (St. Louis, MO, USA). For anesthesia, xylazine and ketamine solutions were purchased from Lloyd Laboratories (Shenandoah, IA, USA) and Fort Dodge Animal Health (Fort Dodge, IA, USA), respectively. All other reagents were analytical grade and obtained from commercial sources.
The dosing solution containing 4 mg/mL each of CyA and the vascular marker FD-70 was prepared by diluting the original solution and the powder, respectively, with Krebs-Henseleit-bicarbonate buffer.
Experimental Protocol
All procedures involving animals used in this study were consistent with the guidelines set by the National Institutes of Health (NIH publication #85-23, revised 1985) and approved by the Institutional Animal Care and Use Committee.
Thirteen adult Sprague-Dawley rats of both sexes were used in this study. The hepatic disposition of CyA was assessed in livers isolated from weight-matched male (n = 6) and female (n = 7) rats. Additionally, the disposition of FD-70 was simultaneously studied as a reference marker for sinusoidal volume and space of Disse because the macromolecule is similar to albumin in terms of MW and volume of distribution (17, 18) and is virtually non-extractable during one single pass through the liver (>99% availability) (17). The procedures for liver isolation and perfusion were similar to those reported by us before (19, 20). Briefly, rats were anesthetized with an i.m. injection of ketamine: xylazine (80:12 mg/kg) mixture, and the common bile duct was cannulated. After cannulation of the portal vein (inlet) and the suprahepatic vena cava (outlet), the liver was isolated and mounted on a temperature (37°C)-controlled perfusion system (MX International, Aurora, Colorado, USA). The perfusate was a Krebs-Henseleit-bicarbonate buffer fortified with 1.2 g/L glucose and 4.75 mg/L of sodium taurocholate and oxygenated with a mixture of oxygen:carbon dioxide (95:5). The perfusate was delivered at a flow rate of 30 mL/min (~3-4 mL/min/g liver weight) in a single pass manner. The livers were allowed to stabilize for 15 min before the start of experiments.
Single bolus doses (1 mg) of CyA and FD-70 were administered simultaneously into the inlet catheter (portal vein), and outlet perfusate samples (~0.5 mL) were collected every 2 seconds for the first 20 seconds and every 5, 15, or 60 seconds thereafter for 15 minutes. Additionally, bile was collected from zero to 15 min. At the end of perfusion, the liver was blotted dry and stored for further analysis. All the samples were stored at -80°C for subsequent measurement of CyA concentrations.
The viability of the liver was assessed through overall macroscopic appearance of the liver, wet liver weights at the end of perfusion, transaminases (ALT and AST) levels in the outlet perfusate at the beginning and end of perfusion, and the flow rate of bile.
Sample Analysis
Liver samples were homogenized in deionized water (1:19) before analysis. The concentrations of CyA in the outlet perfusate, bile, and liver homogenates were then analyzed using an HPLC assay developed recently in our laboratory (21). Briefly, after the addition of internal standard (tamoxifen), samples were extracted into a mixture of ether: methanol (95:5). The organic layer was evaporated and the residue washed with hexane prior to analysis. The separation was achieved using an LC-1 column (70°C) with a mobile phase of methanol:acetonitrile:0.01 M KH2 PO4 (50:25:25, v/v) delivered at a flow rate of 1 mL/min. The eluents were detected at a wavelength of 205 nm.
The concentrations of FD-70 in the samples were analyzed using a previously reported size-exclusion HPLC method (22). Briefly, 20 ml of a trichloroacetic acid solution (20%, w/v) was added to 100 ml of the perfusate sample. The sample was centrifuged and added to autosampler inserts containing 20 ml of 0.3 M NaOH. Separation was achieved on a Hydropore-5-SEC column (Rainin Instrument; Woburn, MA, USA) with a mobile phase of 50 mM phosphate buffer (pH 7.0) delivered at a flow rate of 0.5 ml/min. The samples were detected using a fluorescence detector at excitation and emission wavelengths of 495 and 520 nm, respectively.
Perfusate concentrations of ALT and AST, for assessment of liver viability, were measured by spectrophotometry using commercially available kits.
Pharmacokinetic Analysis
The maximum concentration of CyA and FD-70 in the outlet perfusate samples following the bolus dose administration (CMAX ) and the time to reach this concentration (TMAX ) were obtained by visual examination of the outlet concentration-time profiles. The area under the outlet concentration-time curve (AUC) was calculated using the linear trapezoidal method for the duration of perfusion and extrapolated to infinity using the terminal rate constant (lz ). The cumulative amounts of CyA or FD-70 recovered in the outlet perfusate (
) were estimated using the following equation:
(1)
where t is the specific time during the perfusion or infinity and Q is the perfusate flow rate (30 mL/min). The following equation was used to estimate the total amount of CyA recovered at the end of the 15-min perfusion (
):
(2)
where
and
are the amount of unchanged CyA recovered in the liver tissue at 15 min and the cumulative amount of the drug excreted in bile from zero to 15 min, respectively. For CyA, the terminal rate constant lz , indicative of the net release of CyA from the binding sites in the liver, was also estimated from the terminal slope of logarithm of the amount remaining to be recovered in the perfusate (ARR) versus time plot; ARR was calculated by subtracting
Statistical moment theory (16) was used for estimation of the recovery ratio in the perfusate (F) and mean transit time (MTT) and volume of distribution (V) in the liver for both CyA and the vascular marker FD-70 and for the liver tissue distribution ratio (KL ) of CyA using the following equations:
(3)
(4)
(5)
(6)
where AUMC is the area under the first moment curve. The MTT values were corrected for the transit time through the catheters.
Statistical Analysis
An unpaired two-tailed t test was used for comparison of the pharmacokinetic parameters between the male and female groups. All comparisons were performed at a significance level (a) of 0.05. The data are presented as mean ± SD.
Results
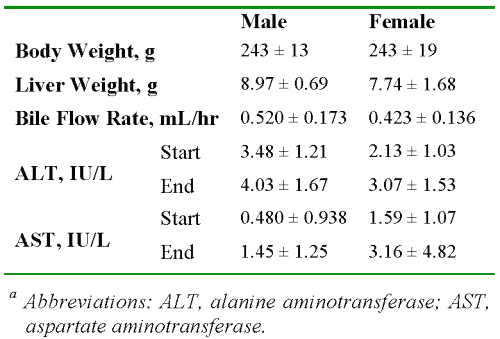
Table 1 presents the data on the characteristics of the livers. There were no significant differences between the males and females in any of the parameters listed in Table 1 (P > 0.05). The wet weights of the livers were less than or equal to 4% of the total body weight at the end of perfusion.
Table 1: The average ± SD values of body and liver weights, bile flow rates, and enzyme levels in male and female rats (n = 6 and 7, respectively).a
Additionally, despite the known hepatotoxicity of CyA (23), which results in reduced bile flow, the observed flow rates in our study (Table 1) were within the range of normal livers. Further, the ALT and AST values for all the livers were very low at the start of the perfusion and remained low for the duration of perfusion (Table 1).
The time courses of the concentrations of CyA in the outlet perfusate of livers from representative male and female rats are presented in Fig. 1, and the corresponding kinetic parameters are listed in Table 2. In both male and female livers, the outlet concentrations rose rapidly, reached a maximum of ~20-25% of dose/mL between 7 to 9 sec, and declined thereafter through an apparent multiexponential process (Fig. 1). In addition to the multiexponential decline during the first 60 sec (Fig. 1, inset), an extremely slow terminal phase was observed in all the livers, which persisted for the entire period of perfusion (Fig. 1).
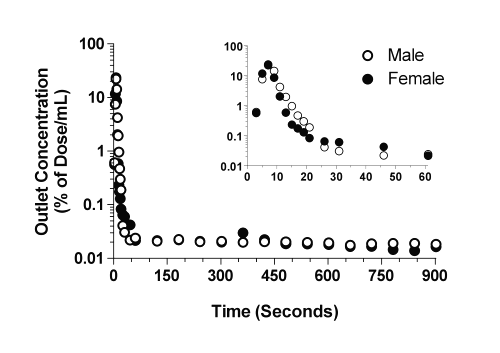
Figure 1: The concentration-time profiles of CyA in the outlet perfusates of representative male and female livers after administration of a bolus 1-mg dose of the drug. The inset depicts the profiles during the initial 60 sec of perfusion.
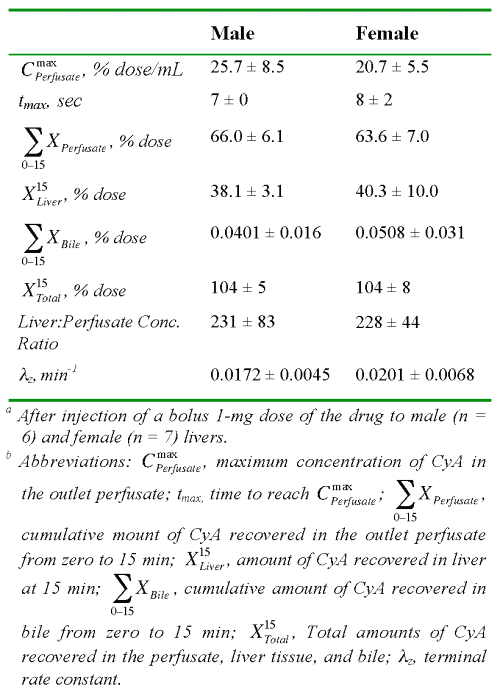
Table 2: The average ± SD values of the observed kinetic parameters of cyclosporine A in isolated perfused rat livers.a,b
An estimated 64 to 66% of the CyA dose was recovered in the outlet perfusate during the 15-min perfusion period (Table 2). Additionally, substantial amounts of the intact drug (~40%) were recovered in the liver tissue at the end of the perfusion (15 min). However, the biliary excretion of the drug during the same period was negligible (Table 2). Therefore, all of the dose could be essentially accounted for by recovery in the outlet and binding to the liver. The substantial binding of CyA to the liver tissue was further demonstrated by a liver:perfusate concentration ratio of ~230 at the end of perfusion (Table 2).
The plots of the amount of CyA remaining to be released into the perfusate versus time are depicted in Fig. 2 for the representative male and female livers. In contrast to the plots in Fig. 1, a clear log-linear terminal portion was observed for all the livers, which was extended from 2 to 15 min (Fig. 2). Therefore, for moment analysis of CyA, the lz values (Table 2) were estimated from ARR plots (Fig. 2) instead of dilution curves (Fig. 1). The harmonic terminal half-life values were 40.3 and 34.5 min for the male and female livers, respectively.
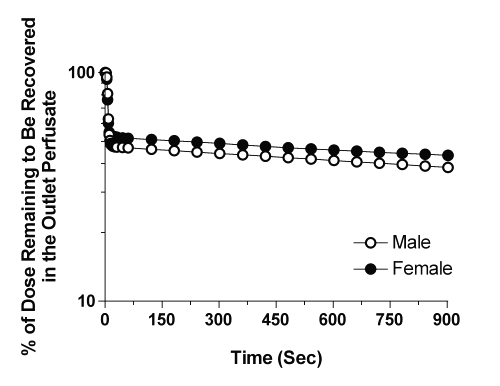
Figure 2: Representative profiles of the amount of CyA remaining to be released from the male and female livers into the outlet perfusate after administration of a bolus 1-mg dose of the drug.
The kinetic parameters estimated from the moment analysis of data for CyA are presented in Table 3.
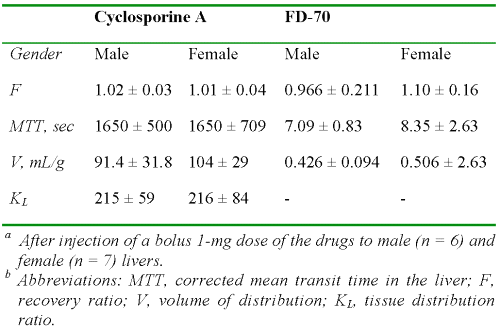
Table 3: The average ± SD values of kinetic parameters of cyclosporine A and FD-70 in isolated perfused rat livers derived from moment analysis of data.a,b
In agreement with the mass balance data after 15 min of perfusion (Table 2), the recovery ratio of CyA estimated from the moment analysis (F) was complete (Table 3). Additionally, the MTT and V values of CyA in the liver were very large (Table 3). Similar kinetic data obtained from the analysis of dilution curves of the marker FD-70 (Fig. 3) are also presented in Table 3.
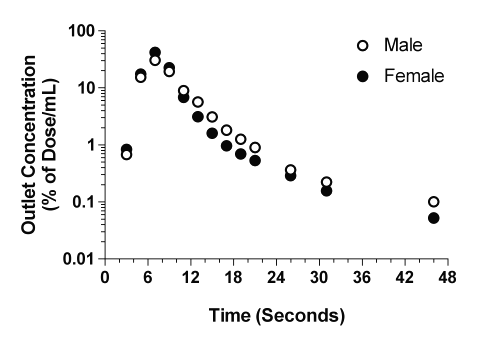
Figure 3: The concentration-time profiles of the marker FD-70 in the outlet perfusates of representative male and female livers after administration of a bolus 1-mg dose of the marker.
As expected, the F of FD-70 was complete and its values of MTT and V were low (Table 3). The tissue distribution ratio (KL) of CyA estimated from the MTT of the drug and the vascular marker using Equation 6 (Table 3) was very close to the observed liver:perfusate concentration ratio at 15 min post perfusion (Table 2).
Discussion
Isolated perfused rat liver model is used either in a recirculating or single-pass mode. Prueksaritanont et al. (15) reported the hepatic disposition of CyA in IPRLs using a recirculating mode after a bolus dose that resulted in an initial CyA concentration of 3.5 μg/mL. They reported an unusual kinetic behavior in that after a rapid decline, the concentrations of CyA in the perfusate started to rise again. Therefore, the kinetic parameters such as terminal half life, total AUC, or clearance could not be estimated. The increase in the perfusate concentrations of CyA at later times during the perfusion was attributed to the release of CyA from its binding sites in the liver tissue due to competition from the formed metabolites (15). Nevertheless, substantial amounts of the intact drug (~80% of the administered dose) were recovered in the liver after 3 hr of perfusion (15), suggesting a slow metabolic process.
Similarly, Deters et al. (23) used a recirculating IPRL model to study the hepatic toxicity of CyA. Using high bolus doses resulting in initial perfusate concentrations of 50-200 mg/mL, these authors showed a rapid uptake of 24-43% of the administered dose. However, the perfusate concentrations remained virtually unchanged during the 120-min perfusion period. Consequently, kinetic parameters could not be estimated from the data.
We became interested in the use of IPRL as a model for delineation of the mechanisms responsible for the higher incidence of allograft rejection in females (24, 25), compared with males, who receive immunosuppressive agents such as cyclosporine. Additionally, it has been reported (26) that the hepatic clearance and volume of distribution of CyA in female patients are ~30% higher than those in males. However, the contributions of these kinetic differences and/or additional dynamic differences to the observed higher organ rejection in females receiving CyA as immunosuppressive therapy are not known. Further, it has been shown that the liver concentrations of CyA at 48 hr after the i.v. injection of 10 mg/kg CyA are substantially lower in female rats, when compared with weight-matched male animals (10). A lower hepatic concentration in females could potentially cause a lower degree of immunosuppression in the liver in case of liver transplantation. However, whether this difference is due to gender differences in the hepatic binding of CyA an/or other confounding in vivo factors is not known. Because of the irregularities in the disposition of CyA in the recirculating IPRL model (15, 23), we investigated the usefulness of a single-pass model. In a preliminary study, we first studied the hepatic kinetics of CyA after constant infusion of the drug at an inlet concentration of ~ 3 mg/mL. However, after 2 hr of perfusion, the concentrations of CyA in the outlet samples were still rising (data not shown), presumably because of significant binding of CyA to the liver tissue. Therefore, the hepatic disposition of CyA was studied in a single-pass IPRL model after a bolus dose, instead of constant infusion, the results of which are presented here (Figs. 1 and 2 and Tables 2 and 3).
After an initial faster decline during the first 60 sec, the dilution curves of CyA showed an unusually slow decline that was sustained for the remaining period of perfusion (Fig. 1). The dilution curves obtained after the bolus injection of drugs or tracers into a single-pass IPRL may be subjected to statistical moment analysis (16). However, such calculations are extremely sensitive to errors in the estimation of the terminal rate constant of the dilution curve. Considering the shallow slope of the CyA profile in the outlet perfusate during the terminal phase coupled with very low CyA concentrations (Fig. 1), the terminal rate constant could not be accurately estimated from the dilution curves in all of our preparations. However, because of an almost complete recovery of the dose in the form of intact drug in the perfusate and the liver tissue (Table 2), we were able to estimate the terminal rate constant from the estimated concentrations in the liver (Fig. 2, Table 2). The very low lz values (Table 2) indicate that the dissociation of CyA from its binding sites in the liver is relatively slow. This is in contrast to the apparently rapid influx of CyA into the liver as evidenced by a substantial accumulation of the dose during one single pass through the liver (Table 2).
Despite substantial accumulation in the liver, negligible amounts of CyA were found in the bile (Table 2). This is in agreement with other studies, recovering in bile only 0.1% of a high dose after 120 min of recirculation (23) and ~2.5% of a low dose after 180 min of recirculation (15) in IPRLs. Nevertheless, in our study, the entire dose was recovered as the intact drug based on both the moment analysis (Table 3) and actual measurements in the perfusate and the liver tissue at the end of perfusion (Table 2), suggesting negligible metabolism of CyA in our IPRL. This is consistent with the very low clearance of CyA observed in vivo in rats, which predicts a true hepatic extraction ratio of ~0.035 (8), and a slow metabolism of the drug relative to its fast uptake in the rat hepatocytes (13). Consequently, the metabolism of a small percentage of the total drug distributed into the liver during one passage through our IPRLs did not affect our overall recovery.
Moment analysis of our data showed an extremely long MTT and large V of CyA in the liver, suggesting that CyA substantially binds to the liver tissue (Table 3). However, our data indicate almost identical binding kinetics for CyA in the livers isolated from the weight-matched male and female rats (Figs. 1, 2, Tables 2, and 3). Therefore, the reported (10) lower accumulation of CyA in the livers of female rats, compared with that in the weight-matched male rats, after in vivo administration could not be due to differences in tissue binding. However, because of the insensitivity to detect subtle differences between the male and female livers in their metabolic activities, existence of such differences cannot be ruled out based on our studies. Indeed, microsomal studies (27) have shown differences in some metabolic pathways of CyA between male and female preparations.
Traditionally, radiolabeled albumin is used as a marker for the volume of sinusoids and space of Disse in multiple indicator dilution studies (28). Because of similarity in the MW and in vivo volume of distribution between albumin and FD-70 and negligible hepatic extraction of both markers (17, 18), we used FD-70 as an alternative to albumin in our studies. The data obtained from the moment analysis of FD-70 (Table 3) supports the use of this macromolecule as an alternative to albumin. This is because the V of FD-70 in the isolated liver (Table 3) is in agreement with the values reported for albumin (28). Additionally, a complete recovery of FD-70 in the perfusate (Table 3) is in line with the characteristics of such a marker. Finally, based on the V of FD-70 (Table 3), the tissue: perfusate concentration ratios of CyA are expected to be close to the KL values (16). Indeed, the KL values (Table 3), which are estimated using the MTT of FD-70, are remarkably close to the experimental concentration ratios (Table 2), suggesting appropriateness of the use of FD-70 as a vascular marker.
In conclusion, our study shows that the tissue binding of cyclosporine A is substantial, slowly reversible, and gender-independent in isolated perfused rat livers. More sophisticated designs allowing multiple measurements of the drug and its metabolites in liver tissue, bile, and perfusate are needed to accurately characterize the kinetics of binding, metabolism, and biliary excretion of CyA in the liver.
Acknowledgement
This study was partially funded by a grant from the Women's Health Research Institute of Amarillo. The authors would like to thank Ms. Katherine Hollenstein for valuable technical assistance and Ms. Ragini Vuppugalla for helpful discussions.
References
Maurer, G., Loosli, H. R., Schreier, E., and Keller, B. Disposition of cyclosporine in several animal species and man. I. Structural elucidation of its metabolites. Drug Metab. Dispos. 12: 120-126, 1984.
Kronbach, T., Fischer, V., and Meyer, U. A. Cyclosporine metabolism in human liver: Identification of a cytochrome P-450III gene family as the major cyclosporine-metabolizing enzyme explains interactions of cyclosporine with other drugs. Clin. Pharmacol. Ther. 43: 630-635, 1988.
Ptachcinski, R. J., Venkataramanan, R., and Burckart, G. J. Clinical pharmacokinetics of cyclosporin. Clin. Pharmacokinet. 11: 107-132, 1986.
Kolars, J. C., Awni, W. M., Merion, R. M., and Watkins, P. B. First-pass metabolism of cyclosporin by the gut. Lancet 338: 1488-1490, 1991.
Kolars, J. C., Stetson, P. L., Rush, B. D., Ruwart, M. J., Schmiedlin-Ren, P., Duell, E. A., Voorhees, J. J., and Watkins, P. B. Cyclosporine metabolism by P450IIIA in rat enterocytes--another determinant of oral bioavailability? Transplantation 53: 596-602, 1992.
Wu, C. Y., Benet, L. Z., Hebert, M. F., Gupta, S. K., Rowland, M., Gomez, D. Y., and Wacher, V. J. Differentiation of absorption and first-pass gut and hepatic metabolism in humans: Studies with cyclosporine. Clin. Pharmacol. Ther. 58: 492-497, 1995.
Fahr, A. Cyclosporin clinical pharmacokinetics. Clin. Pharmacokinet. 24: 472-495, 1993.
Wagner, O., Schreier, E., Heitz, F., and Maurer, G. Tissue distribution, disposition, and metabolism of cyclosporine in rats. Drug Metab. Dispos. 15: 377-383., 1987.
Sangalli, L., Bortolotti, A., Jiritano, L., and Bonati, M. Cyclosporine pharmacokinetics in rats and interspecies comparison in dogs, rabbits, rats, and humans. Drug Metab. Dispos. 16: 749-753, 1988.
Molpeceres, J., Chacon, M., Guzman, M., Aberturas, M. R., and Berges, L. Dependency of cyclosporine tissue distribution and metabolism on the age and gender of rats after a single intravenous dose. Int. J. Pharm. 197: 129-141, 2000.
Wacher, V. J., Liu, T., Roberts, J. P., Ascher, N. L., and Benet, L. Z. Time course of cyclosporine and its metabolites in blood, liver and spleen of naive Lewis rats: Comparison with data obtained in transplanted animals. Biopharm. Drug Dispos. 16: 303-312, 1995.
Bernareggi, A. and Rowland, M. Physiologic modeling of cyclosporine kinetics in rat and man. J. Pharmacokinet. Biopharm. 19: 21-50, 1991.
Prueksaritanont, T., Koike, M., Hoener, B. A., and Benet, L. Z. Transport and metabolism of cyclosporine in isolated rat hepatocytes. The effects of lipids. Biochem. Pharmacol. 43: 1997-2006., 1992.
Smith, J. M., Nemeth, T. L., and McDonald, R. A. Current immunosuppressive agents: efficacy, side effects, and utilization. Pediat. Clin. N. Amer. 50: 1283-1300, 2003.
Prueksaritanont, T., Hoener, B. A., and Benet, L. Z. Effects of low-density lipoprotein and ethinyl estradiol on cyclosporine metabolism in isolated rat liver perfusions. Drug Metab. Dispos. 20: 547-552, 1992.
Kakutani, T., Yamaoka, K., Hashida, M., and Sezaki, H. A new method for assessment of drug disposition in muscle: Application of statistical moment theory to local perfusion systems. J. Pharmacokinet. Biopharm. 13: 609-631, 1985.
Mehvar, R., Robinson, M. A., and Reynolds, J. M. Molecular weight dependent tissue accumulation of dextrans: in vivo studies in rats. J. Pharm. Sci. 83: 1495-1499, 1994.
Lee, H. B. and Blaufox, M. D. Blood volume in the rat. J. Nucl. Med. 26: 72-76, 1985.
Mehvar, R. and Zhang, X. P. Development and application of an isolated perfused rat liver model to study the stimulation and inhibition of tumor necrosis factor-a production ex vivo. Pharm. Res. 19: 47-53, 2002.
Mehvar, R., Reynolds, J. M., Robinson, M. A., and Longstreth, J. A. Enantioselective kinetics of verapamil and norverapamil in isolated perfused rat livers. Pharm. Res. 11: 1815-1819, 1994.
Chimalakonda, A. P., Shah, R. B., and Mehvar, R. High-performance liquid chromatographic analysis of cyclosporin A in rat blood and liver using a commercially available internal standard. J. Chromatogr. B 772: 107-114, 2002.
Mehvar, R. and Shepard, T. L. Molecular weight-dependent pharmacokinetics of fluorescein-labeled dextrans in rats. J. Pharm. Sci. 81: 908-912, 1992.
Deters, M., Strubelt, O., and Younes, M. Reevaluation of cyclosporine induced hepatotoxicity in the isolated perfused rat liver. Toxicology 123: 197-206, 1997.
Esmore, D., Keogh, A., Spratt, P., Jones, B., and Chang, V. Heart transplantation in females. J. Heart Lung Transplant. 10: 335-341, 1991.
Brooks, B. K., Levy, M. F., Jennings, L. W., Abbasoglu, O., Vodapally, M., Goldstein, R. M., Husberg, B. S., Gonwa, T. A., and Klintmalm, G. B. Influence of donor and recipient gender on the outcome of liver transplantation. Transplantation 62: 1784-1787, 1996.
Kahan, B. D., Kramer, W. G., Wideman, C., Flechner, S. M., Lorber, M. I., and Van Buren, C. T. Demographic factors affecting the pharmacokinetics of cyclosporine estimated by radioimmunoassay. Transplantation 41: 459-464, 1986.
Prueksaritanont, T., Correia, M. A., Rettie, A. E., Swinney, D. C., Thomas, P. E., and Benet, L. Z. Cyclosporine metabolism by rat liver microsomes. Evidence for involvement of enzyme(s) other than cytochromes P-450 3A. Drug Metab. Dispos. 21: 730-737., 1993.
Pang, K. S., Lee, W. F., Cherry, W. F., Yuen, V., Accaputo, J., Fayz, S., Schwab, A. J., and Goresky, C. A. Effects of perfusate flow rate on measured blood volume, disse space, intracellular water space, and drug extraction in the perfused rat liver preparation: characterization by the multiple indicator dilution technique. J. Pharmacokinet. Biopharm. 16: 595-631, 1988.
Corresponding Author: Reza Mehvar, School of Pharmacy, Texas Tech Univesity Health Sciences Center, 1300 Coulter, Amarillo, Texas, USA. 79106 reza.mehvar@ttuhsc.edu
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps