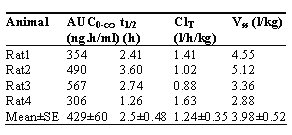
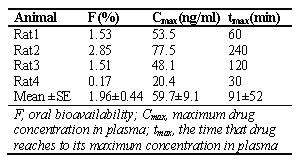
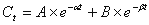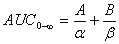J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(3):346-351, 2003
Application of a new high performance liquid chromatography method to the pharmacokinetics of dibudipine in rats.
Shahab Bohlooli, Fariborz Keyhanfar, Saeed Ghiaee, Massoud Mahmoudian1
Razi Institute for Drug Research, Iran University of Medical Sciences, Tehran, IranReceived 17 June 2003, Revised 17 November 2003, Accepted 8 December 2003
PDF version
Abstract
PURPOSE: To develop a HPLC method for assay of dibudipine in biological fluids and to study its pharmacokinetics in the rat. METHODS. HPLC: 2 m l (20 m g/ml) mebudipine as internal standard, 0.2 ml NaOH 1 M and 2 ml ethyl acetate were added to 0.2 ml of rat plasma. The mixture was shaken for 10 min, centrifuged, and the supernatant was dried under nitrogen. The dissolved residue was injected to a C18 analytical column. Mobile phase flowed at 1 ml/min with a composition of methanol--water-acetonitrile (70-25-5). The eluent was monitored at 238 nm. Pharmacokinetic study: plasma samples were collected periodically after intravenous (0.5 mg/kg) or oral (10 mg/kg) administration of dibudipine to rats (n = 4/group). In addition, separate groups of animals were administered 0.5 mg/kg doses of the drug for serial collection of brain, heart, kidney and liver (n = 4/time). The concentration of the drug in tissue or plasma was assayed using the above HPLC method. RESULTS. Calibration curves were linear over a concentration of 10-1000 ng/ml and CV was less than 10%. Dibudipine showed a bi-exponential decline after IV injection in the rats with a t1/2 beta of 2.5 ± 0.5(mean ± SE) hr. Oral bioavailability was low. Distribution of dibudipine to the examined tissues was rapid, and with the exception of the brain, the concentrations of the drug in all tissues were higher than the plasma levels CONCLUSIONS. The HPLC method was simple and convenient. Moreover, it could be applied to investigations of the pharmacokinetics of dibudipine in the rat.
Introduction
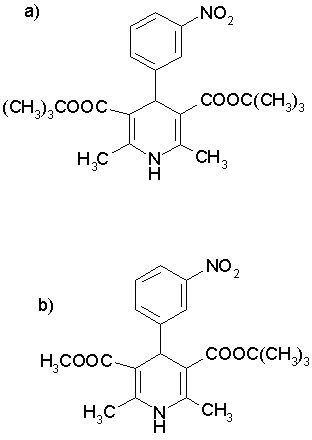
Dibudipine, [bis-t-butyl, -1, 4-dihydro-2, 6-dimethyl-4 - (3-nitrophenyl)-3, 5-pyridine dicarboxylate] is a new 1, 4-dihydropyridine (Figure 1) with calcium channel blocker activity that was synthesized for the first time in our laboratory [1]. In pervious studies, it was shown that dibudipine had comparable pharmacological effect with nifedipine while offering some advantages such as longer biological half-life, longer time to reach peak effect and more vasoselectivity [1, 2]. The HPLC method of mebudipine assay was published previously and dibudipine was used as an internal standard [3]. In this study, high performance liquid chromatography of dibudipine in plasma and tissues is described. Pharmacokinetics of dibudipine after single intravenous injection and oral administration are also studied.
Figure 1: Chemical structure of a) dibudipine and b) mebudipine (internal standard).
Materials And METHODS
Chemicals
Dibudipine and the internal standard (IS, mebudipine) were synthesized in our laboratories [1]. HPLC grade methanol, acetonitrile and ethyl acetate were purchased from Merck (E. Merck, 64271 Darmstadt, Germany). All other reagents were analytical grade or higher.
HPLC system
The HPLC system consisted of a Waters chromatography system (Waters Chromatography Division, Milford, MA, USA) including a 600 controller, a 600 pump, a 486 UV tunable absorbance detector, a 7725i Rheodyne manual injector and Yonglin Data module attached to a PC. An analytical column (Novapack ODS, 5 m m, 4.6 mm ∞ 250 mm) was used for all analysis. The mobile phase consisted of methanol-water-acetonitrile (70-25-5). The chromatography was performed at ambient temperature at flow rate of 1 ml/min and the eluent was monitored at 238 nm.
Standard Solutions
Stock solution of dibudipine and mebudipine were prepared in methanol (1 mg/ml), protected from light and stored at -20o until used. Solutions of 20 and 1 m g/ml were prepared by diluting the stock solution in methanol. The stability of these solutions was compared against a freshly prepared stock solution. Standard curves for plasma were prepared by adding known amounts of dibudipine to pooled blank plasma to yield 10-1000 ng/ml concentrations of the drug.
Sample preparation
To 0.2 ml plasma sample were added 2 m l of IS (mebudipine, 20 m g/ml) and 0.2 ml of 1M NaOH. The solution was mixed for a few seconds. Two ml ethyl acetate was added to the solution, which was subsequently shaken on a horizontal shaker for 10 min followed by centrifugation at 12000 RPM for 10 min. The organic layer was transferred to a clean glass tube and evaporated to dryness under slow stream of nitrogen at 40°C. The resultant residue was reconstituted in 100 m l of mobile phase and 50 m l of final solution was injected into the HPLC. Peak-height ratios of dibudipine: IS in samples were used to calculate dibudipine plasma and tissue concentrations from standards.
Extraction efficacy
The extraction efficiency was calculated by adding known amount of dibudipine (10, 50,100, 500 and 750 ng/ml) to 0.2 ml rat plasma (n =4). Dibudipine was extracted as described above, and the peak heights of dibudipine from spiked plasma samples were compared with the peak heights obtained after direct injection of 50 m l of 10, 50,100, 500 and 750 ng/ml methanolic solutions of dibudipine.
Accuracy and Precision
For the determination of intra- and inter-day accuracy and precision of the assay, aliquots of 0.2 ml of rat plasma were spiked with 2 m l of 20 m g/ml of internal standard and various quantities of dibudipine to yield 10, 50, 100, 500 and 750 ng/ml. Accuracy was expressed as the mean% [(mean measured concentration)/(expected concentration)]×100 [4]. Precision was calculated as inter- and intra-day coefficient of variation [%CV= (SD/mean) ×100] [4].
Pharmacokinetics of dibudipine in Rats
Animal study
Male Sprague-Dawley rats (200±25 g) were used. They were allowed free access to food and water during housing, but were fasted overnight before the study. The drug was dissolved in polyethylene glygol 400 for oral and intravenous administration. The animals were anesthetized by intraperitoneal injection of 25% urethane in isotonic saline (0.5 ml/100g). The external iliac vein was cannulated with catheter and 0.5 ml of heparin (500 IU in saline) was injected before blood collection. In intravenous dosing, 0.5 mg /kg (1 ml/kg) dibudipine was injected into common jugular vein directly via a syringe with 23G needle after a small incision in neck. In oral dosing, 10 mg/kg (5ml/kg) was gavaged into stomach using feeding tube. Blood samples (0.4 ml each time) were periodically collected from external iliac vein. For blood volume compensation, 0.5 ml of saline was injected by catheter. Before dibudipine dosing, 0.2 ml heparin diluted in saline (500u/ml) was injected into jugular vein. Plasma samples were obtained by centrifugation of the blood at 5000 rpm and stored at -20o until analysis. For measuring tissue distribution, 0.5 mg/kg of dibudipine was injected into external iliac vein and animals were killed by severing abdominal aorta at 5, 10, 20 and 30 min after injection. Brain, liver, heart and kidney were immediately removed and washed in saline. One gram of tissues was homogenized in 2 ml saline and centrifuged at 12000 rpm for 30 min. One ml of homogenate supernatant was stored at -20° until analysis.
Pharmacokinetics analysis
Plasma concentrations of dibudipine against time after intravenous administration were fitted to following two-exponential equation using least squares method applying Excel program [5]:
Where A and B are the zero time intercepts of the two components of the two-exponential equation and a and b are the hybrid rate constants of the distribution and elimination phases, respectively [6].
The area under plasma concentration (AUC0-¥) for intravenous injection was calculated as below [6 and 7]:
For oral dosing, AU 0-t was calculated using trapezoidal rule; the highest experimental concentration value was assumed the peak concentration (Cmax ). Time to reach Cmax was denoted as tmax .
The absolute oral bioavailability (F) was calculated as ((AUCpo x dose iv / AUCiv x dosepo )) x 100
Results and Discussion
It was found that liquid phase extraction method yielded the best results in comparison with solid phase extraction method. Ethyl acetate showed good extraction efficiency (>85%) and clean blank chromatograms.
Typical chromatograms of blank plasma, spiked plasma and actual sample obtained from the pharmacokinetic study are shown in Figure 2. Retention time of dibudipine and internal standard were approximately 16 and 7 min, respectively. The total HPLC run time for each sample was about 20 min. There were no interfering peaks in the blank plasma samples. Standard curves prepared for dibudipine in rat plasma were linear over 10 to 1000 ng/ml. The mean (n=6) calibration curve for dibudipine was y= 0.0027x - 0.0241, r2 =0.997 where, y and x are the peak height ratio and concentration (ng/ml), respectively. Detection limit for dibudipine quantitation was 10 ng/ml.
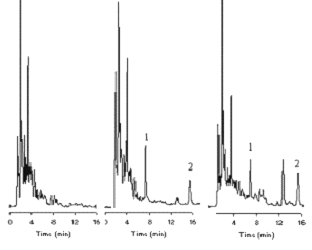
Figure 2: Chromatograms of dibudipine in rat plasma A.) blank rat plasma, B.) sample spiked with 100 ng/ml dibudipine and 100 ng /ml IS, and C.) sample obtained 5 min after IV dose of 0.5 mg/kg of dibudipine. Peaks: 1=mebudipine (IS), 2=dibudipine.
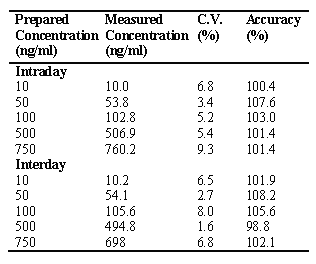
The accuracy of assay was between 90 and 110%, and CV did not exceed 10% (Table 1)
Table 1: Intra and Interday variation of dibudipine assay in rat plasma (n=4).
The method mentioned above was applied to pharmacokinetic study of dibudipine in rats. The plasma concentration-time course and pharmacokinetic parameters of dibudipine after a single iv dose of 0.5 mg/kg are presented in Fig. 3 and Table 2, respectively.
Table 2: Pharmacokinetic parameters of dibudipine following intravenous bolus administration of 0.5 mg/kg dibudipine to rats (n = 4).
The mean plasma concentration was 238 ng/ml at two min after IV administration of 0.5 mg/kg dibudipine. This concentration was declined bi-exponentially to 49 ng after 4 h with a terminal half-life (t1/2â) of 2.5 h (Figure 3, Table 2).
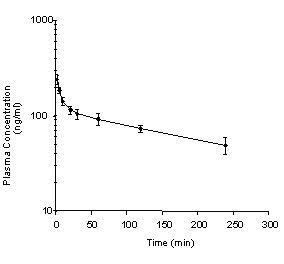
Figure 3: Mean dibudipine plasma concentration-time profile in the rats following IV administration of 0.5 mg/kg of dibudipine. Each point represents the mean +/- SE for four rats.
This half life (2.5 hr) was longer than half lives of other dihydropyridines such as nicardipine (0.1 hr), benidipine (0.5 hr), nisoldipine (0.4 hr), nitrendipine (1.3 hr), felodipine (1.5 hr) and nilvadipine (1.3 hr) [8], but was comparable to that of amlodipine [9]. Clearance of 1.24 l/h/kg for dibudipine (Table 2) was lower than maximum blood flow of rat liver (3.3 to 4.8 l/hr/kg)[10]. Systemic clearance of most dihydropyridines is equal or higher than liver blood flow [8, 11 and 12]. Benidipine [13], nisoldipine [14], nitrendipine [15], felodipine [9], nilvadipine [12] and lacidipine [11] have higher systemic clearance than dibudipine after intravenous administration. Dibudipine plasma concentration declined bi-exponentially (Fig. 3). Plasma concentration of most dihydropyridines has shown bi- or tri-exponential decline [8, 11, 12 , 14 and 15]. Therefore, it appears that the distribution of dibudipine is qualitatively similar to other dihydropyridines. The large Vss of dibudipine (3.98 l/kg, Table 2) indicates substantial distribution of the drug to various tissues consistent with its lipophilicity. Overall, the kinetic data (Table 2) suggest that the relatively long t1/2 of dibudipine is due to its lower systemic clearance coupled with a relatively large Vss, relative to other dihydropyridines.
As shown in Figure 4 and Table 3, after oral administration the plasma concentration-time profiles of unchanged dibudipine showed substantial interindivdual variability.
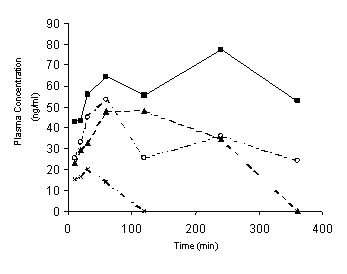
Figure 4: Plasma concentration-time curve of unchanged drug in individual rats after oral administration of 10 mg/kg dibudipine as a solution in PEG 400.
Table 3: Pharmacokinetic parameters of dibudipine following oral administration of 10 mg/kg dibudipine to rats (n = 4).
Mean oral bioavailability of dibudipine was very low (< 2%, Table 3) and similar to oral bioavailability of nilvadipine (4.3%) [16], barnidipine (11-18%) [8], nitrendipine (11%) [15], nisoldipine (3.4%) [14] and benidipine (3.1%) [13] in rats. Despite complete absorption from GI tract, extensive first pass effect is a common property of dihydropyridines like nicardipine, nisoldipine, felodipine, nilvadipine and lacidipine [11]. It seems dibudipine is not an exception to this rule, and with an assumption of complete oral absorption, first pass effect of dibudipine is likely to be extensive in rats.
Dibudipine distribution in several tissues after iv injection of the drug is shown in Figure 5.
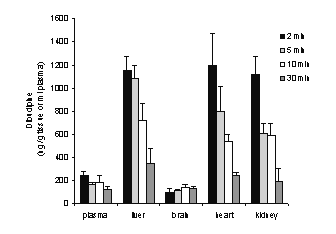
Figure 5: Concentration of unchanged dibudipine in plasma, brain, heart, liver and kidney of the rats at 2, 5, 10 and 30 min after intravenous administration of 0.5 mg/kg dibudipine. Each group represents mean +/- SE for four rats.
Ratios of dibudipine levels in heart, liver and kidney to plasma were constant in various times after the drug administration (2, 5, 10, 30 min), suggesting that these tissues are in the same compartment as plasma (central). In contrast, changes in the brain levels of dibudipine with time were not parallel to its plasma profile. Therefore, it is possible that brain is part of a peripheral compartment in dibudipine disposition [17].
Nevertheless, the concentrations of dibudipine in rat brain were much lower than those in other tissues.
Acknowledgement
The Authors appreciate receiving scientific assistance from Dr Ebrahimi and Dr Motevalian.
References
Mahmoudian M., Mirkhani H., Nehardani Z. and Ghiaee S., Synthesis and biological activity of two new calcium-channel blockers, mebudipine and dibudipine. J Pharm Pharmacol, 49: 1229-1233, 1997.
Mahmoudian M., Mirkhani H., Nehardani Z. and Ghiaee S., Effects of mebudipine and dibudipine, two new calcium-channel blockers, on rat left atrium, rat blood pressure and human internal mammary artery. J Pharm Pharmacol, 51: 617-622, 1999.
Bohlooli S., Keyhanfar F. and Mamoudian M., High performance liquid chromatography of mebudipine: application to pharmacokinetic study. J Pharm Pharm Sci, 4:244-247, 2001.
Sattari S. and Jamali F., High performance liquid chromatographic determination of cyclooxygenase II inhibitor rofecoxib in rat and human plasma. J Pharm Pharmaceut Sci, 3: 312-317, 2000.
Brown AM., A step-by-step guide to non-linear regression analysis of experimental data using a Microsoft Excel spreadsheet. Comput Methods Programs Biomed, 65:191-200, 2001.
Langenbucher F., Handling of computational in vitro/in vivo correlation problems by Microsoft Excel: I. Principles and some general algorithms. Eur J Pharm Biopharm, 53:1-7, 2002.
Langenbucher F., Handling of computational in vitro/in vivo correlation problems by Microsoft Excel II. Distribution functions and moments. Eur J Pharm Biopharm, 55:77-84, 2003.
Teramura T., Watanabe T., Higuchi S. and Hashimoto K., Pharmacokinetics of barnidipine hydrochloride, a new dihydropyridine calcium channel blocker, in the rat, dog and human. Xenobiotica, 25: 1237-1246, 1995.
Stopher D.A., Beresford A.P., Macrae P.V. and Humphrey M.J., The metabolism and pharmacokinetics of amlodipine in humans and animals. J Cardiovasc Pharmacol, 12: S55-S59, 1988.
Grundy JS, Eliot LA, Foster RT., Extrahepatic first-pass metabolism of nifedipine in the rat. Biopharm Drug Dispos. 18: 509-22, 1997.
Pellegatti M., Grossi P., Ayrton J., Evans G.L. and Harker A.J., Absorption, distribution and excretion of lacidipine, a dihydropyridine calcium antagonist, in rat and dog. Xenobiotica, 20: 765-777, 1990.
Tokuma Y., Sekiguchi M., Niwa T. and Noguchi H., Pharmacokinetics of nilvadipine, a new dihydropyridine calcium antagonist, in mice, rats, rabbits and dogs. Xenobiotica, 8: 21-28, 1988.
Kobayashi H, Kobayashi S, Inoue A, Oka T, Nakamizo N., Pharmacokinetic study of benidipine hydrochloride in rats and dogs. Arzneimittelforschung, 38: 1750-3, 1988.
Ahr H.J., Krause H.P., Siefert H.M., Suwelack D. and Weber H., Pharmacokinetics of nisoldipine. I. Absorption, concentration in plasma, and excretion after single administration of [14C]nisoldipine in rats, dogs, monkey, and swine. Arzneimittelforschung, 38: 1093-1098, 1988.
Krause H.P., Ahr H.J., Beermann D., Siefert H.M., Suwelack D. and Weber H., The pharmacokinetics of nitrendipine. I. Absorption, plasma concentrations, and excretion after single administration of [14C]nitrendipine to rats and dogs. Arzneimittelforschung, 38: 1593-1599, 1988.
Tokuma Y, Fujiwara T, Noguchi H., Absorption, distribution and excretion of nilvadipine, a new dihydropyridine calcium antagonist, in rats and dogs. Xenobiotica. 17: 1341-9, 1987.
Gibaldi, M.; Perrier, D.; Pharmacokinetics. Marcel Dekker, New York and Basel, 1982; 1-109.
Corresponding Author: Massoud Mahmoudian, Department of Pharmacology, School of Medicine, Iran University of Medical Sciences, P.O. Box 14155-6183, Tehran,Iran. masmah99@iums.ac.ir
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps