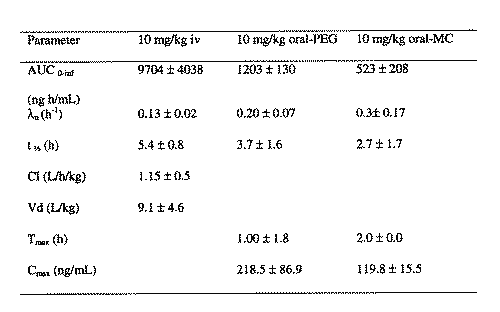J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(2):205-210, 2003
Formulation dependent pharmacokinetics, bioavailability and renal toxicity of a selective cyclooxygenase-1 inhibitor SC-560 in the rat.
Xiao Wei Teng
Department of Pharmaceutial Sciences, College of Pharmacy, Washington State UniversityAbdallalah KM Abu-Mellal
Faculty of Pharmacy, Department of Pharmaceutics, University of Sydney, Sydney, New South Wales, AustraliaNeal M Davies1
Department of Pharmaceutical Sciences, College of Pharmacy, Center for Integrated Biotechnology and Cancer Prevention and Research Center Washington State University, USAReceived 7 May 2003, Revised 4 June 2003, Accepted 4 June 2003
PDF version
Abstract
PURPOSE: To delineate formulation dependent pharmacokinetics and bioavailability of SC-560, a relatively new cycloooxygenase-1 (COX-1) specific inhibitor, in the rat and examine its influence on the renal tubular enzyme, N-acetyl-beta- D -glucosaminidase (NAG), and urinary electrolytes. METHODS: The pharmacokinetics of SC-560 was studied in Sprague-Dawley rats ( n = 5 per group) after a single intravenous (iv) and oral dose (10 mg/kg) in polyethylene glycol (PEG) 600 and a single oral dose (10 mg/kg) in 1 % methylcellulose (MC). Serial blood samples were collected via a catheter inserted in the right jugular vein and serum samples were analysed for SC-560 using reverse phase HPLC. After oral administration of SC-560 in PEG, urine was also collected for 24 h and analysed for urinary sodium, chloride, and potassium as well as NAG. RESULTS: After an iv dose (10 mg/kg) of SC-560, serum AUC, t1/2, CL and Vd were 9704 ± 4038 ng h/mL, 5.4 ± 0.8 h, 1.15 ± 0.46 L/h/kg and 9.1 ± 4.6 L/kg (mean +/- SD, n = 5), respectively. Oral administration of 10 mg/kg SC-560-PEG and MC (n=5 rats) yielded serum AUC, C max, t max and t 1/2 of 1203.4 ± 130.3 and 523 ± 208 ng h/mL, 218.5 ± 86.9 and 119.8 ± 15.5 ng/mL, 1.00 ± 1.8 and 2.0± 0 h, 3.7 ± 1.6 and 2.7 ± 1.7 h (mean +/- SD, n = 5), respectively. A single oral dose 10 mg/kg of SC-560 in PEG resulted in an increase in NAG excretion in urine and a reduction in 0-24 h urinary sodium, potassium, and chloride excretion. CONCLUSIONS: SC-560 extensively distributes into rat tissues, and has a CL approaching hepatic plasma flow. The drug displays low <15% and formulation dependent bioavailability after oral administration and demonstrates kidney toxicity.
Introduction
The discovery of two isoforms of cyclooxygenase (COX) has led to the development of selective inhibitors of each isoform. SC-560 (5-(4-chlorophenyl)-1-(4-methoxyphenyl)-3-(trifluoromethyl)-1H-pyrazole) (Fig. 1) is a diaryl hetorocycle selective inhibitor of COX-1 (1).
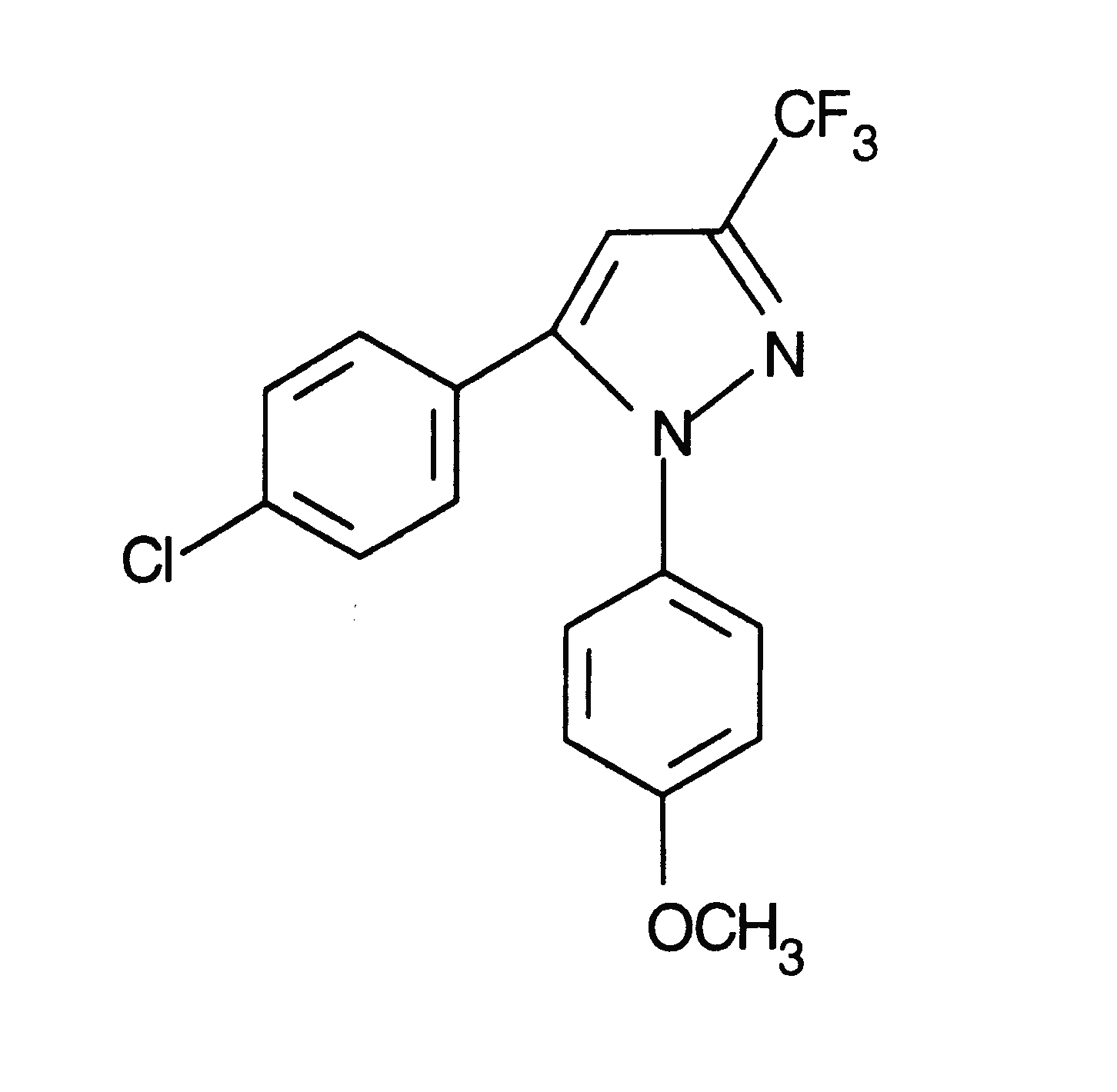
Figure 1: The Chemical Structure of SC-560.
The fifty percent inhibitory concentrations (IC50 ) values for SC-560 are 9 nM and 6.4 m M for COX-1 and COX-2, respectively using human recombinant enzymes (1). SC-560 has been suggested to be orally active in the rat, as 10 mg/kg completely abolishes the ionophore production of thromboxane B2 in whole blood (2). SC-560 is being extensively used in pharmacological and toxicological studies as a selective COX-1 inhibitor (3, 4, 5, 6, 7). However, the use of this compound in previous investigations has been undertaken with a complete absence of knowledge of its pharmacokinetics. SC-560 is structurally related to celecoxib, a selective inhibitor of COX-2. Similar to celecoxib, SC-560 has poor aqueous solubility and thus its pharmacokinetics and in particular, its oral bioavailability may be an important determinant of its onset and duration of action (8). Interestingly, some investigators administer this compound in hydroxypropylcellulose, 5% gum arabic, 0.25% or 1% methylcellulose, without the knowledge of the impact of these dosage forms on the extent of oral bioavailability or pharmacodynamic effects (4, 5, 6). In addition, there is no information on whether the experimental disease states that SC-560 is administered in can also affect the pharmacokinetics of this compound.
In order to elucidate and understand pharmacokinetic / pharmacodynamic relationships of this COX-1 inhibitor, knowledge of its pharmacokinetics are of considerable importance. To our knowledge, no study has been published characterizing the pharmacokinetics of SC-560 and the oral activity of this drug on the renal tubular enzyme N-acetyl-beta- D -glucosaminidase (NAG) and urinary electrolytes. The present paper describes the pharmacokinetics of SC-560 in rat after oral and intravenous administration. In addition, the impact of oral SC-560 on renal tubular enzyme (NAG) and urinary electrolytes was evaluated.
Methods and Materials
Chemicals
SC-560 was purchased from Cayman Chemicals (Ann Arbor, MI. USA). HPLC grade methanol, acetonitrile, and water were purchased from J. T. Baker (Phillipsburg, NJ, USA). Testosterone 17-propionate, methylcellulose, blood urea nitrogen (BUN), and creatinine kits were obtained from Sigma Chemicals (St. Louis, MO, USA). Polyethylene glycol (PEG) 600 was purchased from Union Carbide Chemicals (Danbury, CT, USA). Rats were obtained from Charles River Laboratories. NAG kits were purchased from Boehringer Mannheim Biochemica (Shionagi & Co. Ltd. Osaka, Japan).
Animals and Surgical Procedures
Male Sprague-Dawley rats (170-200g) were used in these studies. Animal ethics approval was obtained from the Institutional Animal Care and Use Committee at Washington State University. Rats were housed in temperature-controlled rooms with a 12 h light/dark cycle. The day before the experiment, the right jugular veins of the rats were catheterized with sterile silastic cannulae (Dow Corning, Midland, MI) under halothane anaesthesia. The animals were transferred to metabolic cages and were fasted 12 h and allowed free access to water before and during the pharmacokinetic study.
Dosing and Sampling Collection
On the morning of study, SC-560 was weighed and dissolved in PEG 600 or suspended in 1% methylcellulose (MC) (~0.5 mL). The formulations were used for dosing rats orally, via gavage needle, or intravenously (iv). Rats received 10 mg/kg (N = 5) of SC-560 (PEG) iv. Five rats received oral doses of 10 mg/kg of SC-560 in PEG or MC. Saline was used to flush the cannulae immediately after injection of the drug (~0.25 mL) and after each collection of blood. Blood (250 m L) was collected pre-dose through the catheter and at 2, 10 min, and 0.5, 1, 2, 4, 8, 12, and 24 h after iv dosing and at 0.25, 0.5, 1, 2, 4, 8, 12, and 24 h after oral dosing. In a separate study, five rats received ~0.5 mL of vehicle or 10 mg/kg of SC-560 in PEG orally and urine was collected for 24 hours and volume measured. All specimens were kept at -70°C until analysis. Urine was thawed at room temperature and vortexed for 30 seconds and a sample of 0.05 mL was used in each analysis according to manufacturer's instructions. 3-Cresolsulfonphthalenyl-N-acetyl-b-D-glucosaminide, sodium salt is hydrolysed by NAG with the release of 3-cresolsulfonphthalein, sodium salt (3-cresol purple), which is measured photometrically at 580 nm.
Assay
A validated HPLC procedure was used for assay of SC-560 in rat serum (9). To serum samples (0.1 ml) was added 50 m l of internal standard solution of testosterone 17-propionate, (10 m g/ml) and 1 mL ice-cold acetonitrile. The mixture was vortexed for 1 min (Vortex Genie-2, VWR Scientific, West Chester, PA, USA), and centrifuged at 15000 rpm at 4°C for 5 min (Beckman Microfuge, Beckman Coulter, Inc., Fullerton, CA, USA USA). The supernatant was collected and evaporated to dryness using a Heto Vac concentrator (Heto-Holten, DK-3450 Allerød, Denmark). The residue was reconstituted with 100 ml of 70% methanol (v/v), vortexed for 1 min and centrifuged at 8000 rpm at 4°C for 5 min, and 40 m l of the supernatant was injected onto the column. The HPLC system used was a Shimadzu HPLC (Kyoto, Japan), consisting of an LC-10AT VP pump, an SIL-10AF auto injector, an SPD-M10A VP spectrophotometric diodearray detector, and an SCL-10A VP system controller. Data collection and integration were accomplished using Shimadzu EZ Start 7.1.1 SP1 (Kyoto, Japan).The analytical column used was Beckman ultrasphere octyl column (150 × 2 mm I.D., 5-m particle size, Beckman Instrument, Fullerton, CA, USA) equipped with a pre-column (7.5 × 2 mm I.D., 5 m) of the same packing material. The mobile phase consisted of methanol and water (7:3, v/v), filtered and degassed under reduced pressure, prior to use. Separation was carried out isocratically at ambient temperature (25 ± 1°C), and a flow rate of 0.25 mL/min, with UV detection at 240 nm. For all runs, quality control samples were incorporated to ensure integrity of the results. Standard curves were linear, and bias and precision data were less than 10% at high (5000 ng/mL) and low (20 ng/mL) drug concentrations. The accuracy was estimated based on the mean percentage error of measured concentration to the actual concentration (10).
Pharmacokinetic Analysis
The elimination rate constant (λn) was estimated by linear regression of the serum concentrations in the log-linear terminal phase. Peak concentrations of SC-560 in the serum (Cmax ) and the corresponding Tmax were estimated for each rat from the serum concentration profile using WinNonlin® (version 1.0). In order to estimate serum concentrations (C0 ) immediately after injection of iv SC-560, compartmental models were fitted to the serum concentration versus time data using WinNonlin® (version 1.0). The estimated C0 was then used in conjunction with the actual measured serum concentrations to determine the area under the plasma concentration-time curve (AUC). The AUC0∞· was calculated using the combined log-linear trapezoidal rule for data from time of dosing to the last measured concentration, plus the quotient of the last measured concentration divided by λn. Non-compartmental pharmacokinetic methods were used to calculate clearance (CL) and volume of distribution (Vd) after iv dosing. The oral bioavailability (F) was calculated as follows:
Urine Electrolytes
Urine samples (0.05 mL) were assayed for potassium, chloride and sodium using commercially prepared reagents obtained from Beckman Coulter Inc. a Beckman, Synchrome, and EI-ISE, Electrolite system (Beckman Coulter Inc. Brea, CA USA).
Statistical Analysis
Statistical analysis of the pharmacokinetic data was performed using Student's t-test for unpaired samples. Microsoft Excel 97 (Microsoft, Redmond, WA, USA.). A p value of less than 0.05 was considered statistically significant. Statistical analysis was performed using Summary data were expressed as mean ± standard deviation (SD).
Results
Multi-exponential decline was noted in the serum concentration versus time profiles after iv administration (Fig. 2).
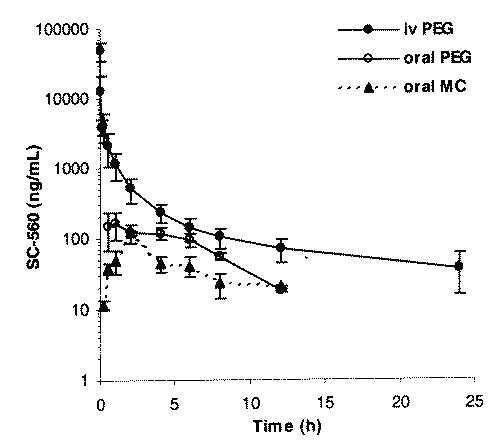
Figure 2: Concentration time profile of SC-560 following iv and oral administration of SC-560 (10 mg/kg) to rats (mean ± SD, n = 5).
Non-compartmental analysis was employed in all pharmacokinetic analysis. Mean extrapolated AUC after each iv and oral dose was less than 20%. The mean V d of SC-560 was 9.1 ± 4.6 L/kg after iv administration. The mean t1/2 of SC-560 was ~5 h (Table 1). After oral dosing, of the PEG formulation in each of the rats there were measurable amounts of drug in serum at time (15 min) of the first post-dose blood sample (Fig. 2). The tmax ranged from 0.5 to 4 h, and Cmax ranged from 150 to 316 ng/mL (Fig. 2, Table 1). After oral dosing, of the MC formulation in each of the rats there were measurable amounts of drug in serum at time (15 min) of the first post-dose blood sample (Fig. 2). The mean tmax was 2 h, and Cmax ranged from 102 to 132 ng/mL (Fig. 2, Table 1). Comparing the AUC of the iv dose with that obtained after oral dosing of PEG and MC, the mean oral bioavailability of SC-560 in the rat (Table 1) was estimated to be 15 and 5%, respectively.
Table 1: Pharmacokinetics data of SC-560 after iv and oral administration in rats (mean ± SD, n=5).
There were no significant differences observed in the terminal phase t1/2 between the oral and iv doses. The urinary NAG amount increased after a single oral dose of SC-560 10 mg/kg in PEG compared to the control group over 24 h (Fig. 3).
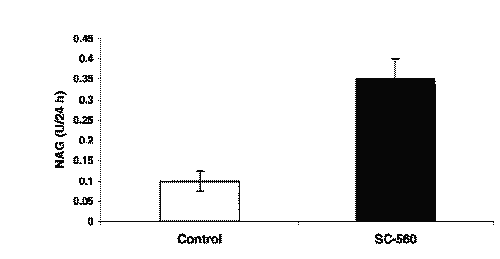
Figure 3: NAG Activity in International Units Per Total Urine Volume Collected over 24 hours following a single oral dose of SC-560 in PEG 10 mg/kg (N=5, Mean +/- SD).
SC-560 also produced a significant reduction in urinary sodium, chloride and potassium excretion over 24 h compared to controls (Fig. 4). No significant changes in serum creatinine or BUN concentration were apparent between SC-560 treated and control group. (data not shown)
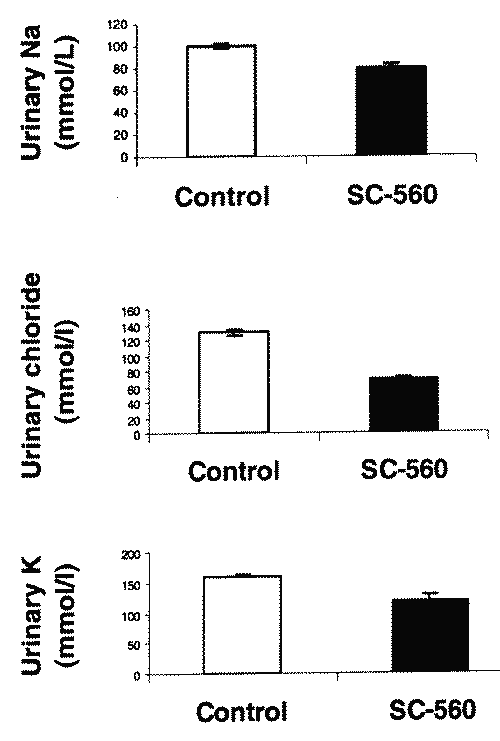
Figure 4: Urinary sodium (top), chloride (middle) and potassium (bottom) after a single oral 10 mg/kg dose of with SC-560 in PEG in μMol/min (N=5; Mean +/- SD).
DISCUSSION
As suggested by its large Vd (mean 9.1 ± 4.6 L/kg), which greatly exceeds body water composition, SC-560 is highly distributed into the rat tissues. The pharmacokinetic parameters of SC-560 in the rat are similar to celecoxib, which similarly possess a large Vd (11). SC-560 is a very lipophilic drug, and is sparingly soluble in aqueous buffers. This physiochemical property is likely a contributor to its large Vd, as it would promote uptake of drug into tissues of high lipid content, such as adipose and brain.
Causes of low oral bioavailability include high first-pass metabolism by the liver and/or incomplete transfer of drug from the gastrointestinal tract to the portal vein. In the rat, mean hepatic blood flow is 3.3 L/h/kg with a hematocrit of 0.48; this yields a mean hepatic plasma flow of ~1.74 L/h/kg (12). Therefore, the serum clearance of SC-560 after 10 mg/kg of 1.15 ± 0.46 L/h/kg is approaching the mean hepatic plasma flow in the rat Some involvement of incomplete transfer from the gastrointestinal tract is apparent as SC-560 bioavailability in PEG increased 3 fold compared to the MC suspension.
Previous pharmacodynamic and pharmacological studies of SC-560 in vivo have administered this compound intravenously and orally in various formulations without any knowledge of its pharmacokinetics (1, 2, 3, 4, 5, 6, 7). The current studies suggest that SC-560 undergoes route of administration and formulation dependent pharmacokinetics.
Furthermore, inflammation may reduce clearance of drugs cleared in the liver (13) and SC-560 administration in animal models of disease that involve the liver (i.e. cirrhosis) may demonstrate altered pharmacokinetics (14, 15, 16).
In agreement with previous studies, we found that SC-560 is orally active (1, 5). SC-560 induced a significant increase in NAG activity per total urine volume collected over 24 h. NAG is a lysosomal enzyme found in the renal tubules with a molecular weight of 150,000 daltons and thus its large size prevents it from reaching the nephron lumen because it cannot undergo glomerular filtration. NAG has been suggested to be a specific marker of renal tubular damage (17, 18). Furthermore, a significant decrease in urinary sodium, potassium, and chloride excretion was evident. The decrease in electrolyte excretion may be related to constitutive COX-1 inhibition in the kidney. Interestingly, treatment with SC-560 did not produce any significant changes in either BUN level or serum creatinine after a single dose. We have found that serum creatinine and BUN are relatively insensitive measures of renal toxicity in the rat after administration of single therapeutic doses of non-steroidal anti-inflammatory drugs and various other COX inhibitors (19). The only other study on the pharmacodynamic effects of SC-560 on renal function was undertaken in rats with cirrhosis and ascites, which also demonstrated a significant decrease in urinary sodium and a significant reduction in glomerular filtration rate, renal plasma flow, and renal prostaglandin E2 after a 20 mg/kg iv dose (16). It is noteworthy that in cirrhotic rats the effects of selective COX-1 inhibition on furosemide-induced diuretic and naturetic response with SC-560 were dose-dependent (16).
In summary, SC-560 is a lipophilic drug characterized by a clearance approaching hepatic plasma flow, a high Vd and a low and formulation dependent bioavailability. A single oral dose of 10 mg/kg demonstrated an ability to reduce urinary electrolytes and induced renal tubular damage, as measured by an increase in NAG. This study is the first report regarding the pharmacokinetics and bioavailability of SC-560 and its effects on NAG excretion and renal electrolytes in animals without disease. Further studies characterizing the pharmacokinetic/pharmacodynamic relationships of selective COX-1 inhibition are currently in progress.
ACKNOWLEDGMENTS
American Cancer Society Institutional Research Grant to NMD.
References
Smith, C. J., Zhang, Y., Koboldt, C. M., Muhammad, J., Zweifel, B. S., Shaffer, A., Talley, J. J., Masferrer, J. L., Seibert, K., Isakson, P, C., Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc Natl Acad Sci U S A, 95:13313-13318, 1998.
Masferrer, J.L., Zweifel, B.S., Manning, P.T., Selective inhibition of inducible cyclooxygenase2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci U S A, 91: 3228-3232, 1994.
Wallace, J.L., McKnight, W., Reuter, B.K, Vergnolle, N., NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology 119: 706-714, 2000.
Sawdy, R.J., Slater, D.M., Dennes, W.J., Sullivan, M.H., Bennett, P.R., The roles of the cyclo-oxygenases types one and two in prostaglandin synthesis in human fetal membranes at term. Placenta 21: 54-57, 2000.
Gretzer, B., Maricic, N., Respondek, M., Schuligoi, R., Peskar, B.M., Effects of specific inhibition of cyclo-oxygenase-1 and cyclo-oxygenase-2 in the rat stomach with normal mucosa and after acid challenge. Br J Pharmacol 132: 1565-1573, 2000.
Loftin, C.D., Trivedi, D.B., Langenbach. R., Cyclooxygenase-1-selective inhibition prolongs gestation in mice without adverse effects on the ductus arteriosus. J Clin Invest 110: 549-557, 2002.
Tanaka, A., Hase, S., Miyazawa, T., Takeuchi, K., Up-regulation of cyclooxygenase-2 by inhibition of cyclooxygenase-1: a key to non-steroidal anti-inflammatory drug-induced intestinal damage. J Pharmacol Exp Ther 300: 754-761, 2002.
Davies, N.M., McLachlan, A.J., Day, R.O., Williams, K.M., Clinical pharmacokinetics and pharmacodynamics of celecoxib: a selective cyclo-oxygenase-2 inhibitor. Clin Pharmacokinet 38: 225-242, 2000.
Teng, X.W., Davies, N.M., High-Performance Liquid Chromatographic Analysis of a Selective Cyclooxygenase-1 Inhibitor SC-560 in Rat Serum. J Pharm Biomed Anal In press 2003.
Shah, V.P., Midha, K.K., Dighe, S., McGilveray, I.J., Skelly, J.P., Yacobi, A., Layloff, T., Viswanathan, C.T., Cook, C.E., McDowall, R.D., Pittman, K.A., Spector, S., Analytical methods validation: bioavailability, bioequivalence and pharmacokinetic studies. Conference report. Eur J Drug Metab Pharmacokinet. 16: 249-255, 1991.
Guirguis, M.S., Sattari, S., Jamali, F., Pharmacokinetics of celecoxib in the presence and absence of interferon-induced acute inflammation in the rat: application of a novel HPLC assay. J Pharm Pharmaceut Sci 4: 1-6, 2001.
Davies, B., Morris, T. Physiological parameters in laboratory animals and humans. Pharm Res 10: 1093-1095, 1993.
Piquette-Miller, M., Jamali, F., Selective Effect of Adjuvant arthritis on the disposition of propranolol enantiomers in rats detected using a stereospecific HPLC assay. Pharm Res 10: 294-299, 1993.
Johnson, P.M., Vogt, S.K., Burney, M.W., Muglia, L.J., COX-2 inhibition attenuates anorexia during systemic inflammation without impairing cytokine production. Am J Physiol Endocrinol Metab 282: E650-E656, 2002.
Rudnick, D.A., Perlmutter, D.H., Muglia, L.J., Prostaglandins are required for CREB activation and cellular proliferation during liver regeneration. Proc Natl Acad Sci U S A 98: 8885-8890, 2001.
Lopez-Parra, M., Claria, J., Planaguma, A., Titos, E., Masferrer, J.L, Woerner, B.M, Koki, A.T., Jimenez, W., Altuna, R., Arroyo, V., Rivera, F., Rodes, J., Cyclooxygenase-1 derived prostaglandins are involved in the maintenance of renal function in rats with cirrhosis and ascites. Br J Pharmacol 135: 891-900, 2002.
Porter, G.A., Norton, T.L., Legg, V., Using urinary biomarkers to evaluate renal effects of a COX-2 NSAID in volunteers. Renal Failure 21: 311-317, 1999.
Meyer, B.H., The use of renal enzymes as early indication of renal toxicity after multiple-dose administration of aspirin. South Af Med J 63: 687-688, 1983.
Abu-Mellal, A.M., Investigations of renal and gastrointestinal toxicity of selected anti-inflammatory drugs in a rat model. MSc Thesis University of Sydney 2002.
Corresponding Author: Neal M Davies, College of Pharmacy, Department of Pharmaceutical Sciences, Washington State University, Pullman, Washington, 99164-6534, USA. ndavies@wsu.edu
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps