J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(2):211-214, 2003
Drug registration application in China.
Li Hui Zhen1
International Medical and Commercial College, China Pharmaceutical University, NanJing, ChinaReceived 14 April 2003, Revised 11 May 2003, Accepted 14 June 2003
PDF version
Abstract
PURPOSE: This article is used to give a brief overview for people who would like to submit a drug registration application in China, or for those who would like to get a broader international drug registration perspective. METHODS. This paper concretely describes the new items in the current drug registration application through introducing following contents: qualification of the applicant, registration classification or type, the procedures for drug registration application review, the intellectual property rights concerning the pharmaceutical (drug substance and product), the process for submitting a drug registration application and the materials required in application for registration. RESULTS. From the paper, we have a comprehensive knowledge of drug registration application in China. CONCLUSIONS. The current Provision for Drug Registration is more reasonable and suitable for China's entry into WTO and further guarantee that safe and effective drugs are available to Chinese people.
Introduction
The current Provision for Drug Registration was promulgated on October 30, 2002, and put into force on December 1, 2002. Compared with the former regulations, the current Provision for Drug Registration is more reasonable and suitable, particularly with regard to China's entry into World Trade Organization (WTO). This paper introduces and discusses the salient features of drug registration applications regulated by the current Provision for Drug Registration
The Regulation on the Qualification of Drug Registration Applicant [1]
The qualification of drug registration applicant was not subject to regulation according to the former New Drug Approval Regulation. This resulted in applications for a new drug approval by anyone. However the research and development of new drugs is now a high-cost and high-risk industry, many individuals may not be qualified to be drug registration applicants when their risk-resistance ability, limited funds and personal experiences are taken into account. In addition, the facts proved many individuals would alter, forge and counterfeit experimental or trial data, materials and reports in order to obtain the Certificate of Drug Approval in the past decades. Moreover, many of them did not produce the new drugs for the reason of being short of funds after obtaining the Certificate of Drug Approval. They usually transferred the new drug technology to other person or entity time after time, no considering whether the transferees can produce the new drugs in a GMP-compliant, manufacturing facilities. This situation finally lead to a new drug may be produced in a low quality by many manufacturing enterprises. [2] [3]
The current Provision for Drug Registration clearly stipulates the qualifications required of drug registration applicants. The drug registration applicant is an entity who submits the drug registration application, assumes corresponding legal obligation and owns the Certificate of Drug Approval once the application is approved. The applicant is usually a corporation, pharmaceutical firm, government agency or scientific institutions. In Chinese territory, the applicant should be a legal entity. If an applicant with an address outside of Chinese territory wishes to apply for import drug registration, he should depend on his branch located in China or China's authorized agency. In addition, the assigned person taking charge of the drug registration application should be a person who has sufficient knowledge of his field at locally and internationally and is proficient in administration of the regulations on drug registration.
The Classification of Drug Registration Application
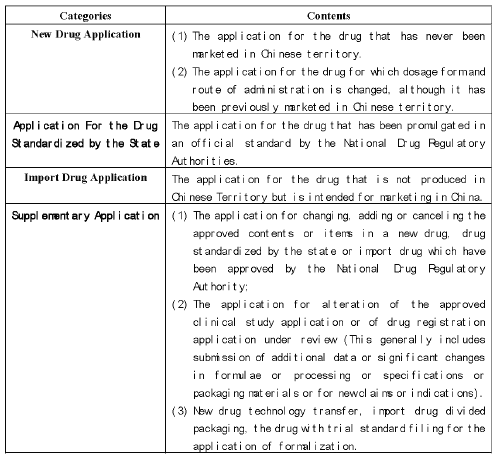
There are four categories of drug registration application, including
New Drug Application,
Application for the drug standardized by the state (which used to be called generic drug), This are roughly equivalent to Abbreviated New Drug Applications in U.S. or Submission in Canada (ANDA or ANDS)
Import drug application and
Supplementary application
(The detailed information on these applications is listed in chart 1).
Chart 1: The Classification of the Drug Registration Application.
An applicant in Chinese territory can register a new drug application, an application for the drug standardized by the state, and a supplementary application. An import drug application and supplementary application are appropriate to the applicant beyond Chinese territory.
The current Provision for Drug Registration defines "A New Drug" as a drug that has never been marketed in Chinese territory. This differs from the old Regulation that defined New Drugs as those produced in China for the first time, According to this new definition, Chinese manufacturers, who already produce imported drugs on the Chinese market, should submit the application according to the requirements for "the drug standardized by the state", thereby solving the problem of a too general definition such that the "new drug" was not "new" in the real sense.
The Procedures for the Review of Drug Registration Application [4]
Generally speaking, the procedures for the review of drug registration application cover the following steps:
After receiving the drug registration application, the Provincial Drug Administration Authorities (PDAAs) should organize the works of the formal review of submitted materials, on-site examination and sampling. The formal review aims to guarantee the content and format of the submitted materials is in line with the requirements and all the required materials have been submitted. After formal review, the PDAAs send the qualified applications to the State Drug Administration (SDA) for further review. The import drug registration application should be directly submitted to SDA by the applicant.
SDA's Department of Drug Registration carefully reviews the completeness of the submitted materials, files the qualified applications and transmits all the materials of qualified applications to the Center for Drug Evaluation directly attracted to SDA.
The mission of SDA's Center for Drug Evaluation (CDE) is to assure all the drugs available to Chinese people are safe and effective, which is roughly equivalent to that of SDA's Center for Drug Evaluation and Research in USA. CDE organizes drug evaluation experts to review the application and determine whether the safety and effectiveness information submitted for a new drug are adequate for manufacturing and marketing approval. The drug evaluation experts are selected from such field as medical treatment, research, laboratory examination and teaching; proficient in the knowledge of their fields locally and internationally and with senior technical titles. The review usually includes chemistry review, pharmacology/toxicology review, statistical review, safety review, clinical review, biopharmaceutical review and labeling review [9].
Carefully considering the recommendations and review results of CDE, SDA makes a decision whether or not the drug registration application can be approved and issues the Certificate of Drug Approval and drug approval number to the qualified applicant.
It is necessary to inspect the drug sample and review the drug's proposed standard during the review. The drug's proposed standard review means to see whether the drug's inspection method is feasible and scientific and whether the proposed index can control the drug quality. The National Institute for the Control of Pharmaceutical and Biological Products (NICPBP) is responsible for the drug sample inspection and drug's proposed standard review of import drugs and biological products. The Provincial Institutes are responsible for the drug sample inspection and drug's proposed standard review of new drugs, drugs standardized by the state and supplementary applications.
The Protection of the undisclosed trial data in drug application [1]
The state protects the undisclosed trial and other data obtained by the producer or distributor who have been granted the production or distribution license of the drugs containing a new chemical entity. It is prohibited to use the undisclosed trial and other proprietary data for illegal commercial purposes. Within six years after the drug manufacturer or distributor is granted the production or distribution license of the drug containing a new chemical entity, the drug regulatory authorities will not approve use of the above-mentioned proprietary data to apply for the production and distribution of the new chemical entity unless with the permission of the licensee.
This is the commitment required to be honored in association with China's membership in WTO.
Intellectual Property Right on Pharmaceutical [1]
With China's accession to WTO, patent protection for pharmaceuticals assumes great importance within domestic and foreign pharmaceutical community. Thus, the current Provision for Drug Registration has added some new items on intellectual property right protection for pharmaceuticals.
The applicant should submit the patent information and ownership certificate for the drug submitted for registration, or for the formula and technology used in the research and manufacture of submitted drug, the guarantee of not constituting an infringement, and the promise of assuming all infringement responsibilities.
DA allows the applicant to submit a drug registration application under the patent protection, within the two years of the patent deadline. This assists generic manufacturers to put their products on the market as soon as patent expiry occurs.
How to submit a drug registration application
When applying for drug registration the applicant should submit the application to the Provincial Drug Administrative Authority in his province, along with the related material and drug sample for his province. When applying for import drug registration, the applicant should submit the application directly to the State Drug Administration.
When the new drug co-applicant comprise two or more organizations and these have addresses that are not in the same province: (1) If only one of them is a pharmaceutical manufacturing enterprise, the application should be submitted to the Provincial Drug Administrative Authority where the pharmaceutical enterprise is located; (2) If two or more of them are pharmaceutical manufacturing enterprises, the application should be submitted to the provincial drug administrative authority where the pharmaceutical manufacturing enterprise manufacturing the preparation is located; (3) If none of them is pharmaceutical manufacturing enterprise, the application should be submitted to the Provincial Drug Administrative Authority where the final drug product will be manufactured.
What materials should be submitted for application [5]
When applying for a chemical drug import registration, the foreign enterprise should submit the required materials including summary, pharmaceutical development studies, animal pharmacology/toxicology studies and clinical studies. (The detailed information is listed in chart 2).
Chart 2: Materials required for Submission.
In addition, the applicant should file the following dossiers: (1) The verified documents for market access to the country of origin of the drug, approved or recognized by the government of that country or region if the drug has been marketed If not, the documents can be submitted to SDA along with clinical study report after the clinical study has been done in China; (2) The copy of the Certificate for Foreign Firm's Branch Permanently Located in China when the applicant's branch located in China filing the application; or the copy of authorization, notarization and the Business License of the China's agency when the China's agency is authorized to submit the application; (3) The patent information and ownership certificate of the drug submitted for registration, or of the formula and technology used in manufacture of the drug, and the guarantee of not constituting an infringement.
Copies of all the above materials and dossiers should be provided prepared or translated into Chinese.
CONCLUSION
From the above introduction, we can conclude the current Provision for Drug Registration has added many new items such as the regulation on the qualification of drug registration applicant, the classification of drug registration application, the protection of the undisclosed trial data and etc. Consequently, the new provision is more reasonable and suitable for China's entry into WTO and further guarantee that safe and effective drugs are available to the Chinese people.
References
35The Provision for Drug Registration. Available from: http:// www.sda.gov.cn/webportal/portal.po?UID=DWV1_WOUID_URL_53384&TOC=CO LUMN_53384&OBJ=17206397
the Drug Administration Law. Available from: http:// www.sda.gov.cn/webportal/portal.po?UID=DWV1_WOUID_URL_68210&TOC=CO LUMN_68210&OBJ=21845
Zhaoming, The Suggestion on the Procedures and Mechanism of New Drug Review. Chinese Journal of New Drug 2002, Vol.11 No.5:345-349
Yezhuguang. The Opinion on the New Drug Review. Chinese Journal of New Drug 2002, Vol.11 No.5:265-268
The Registration Classification and Required Submitted Materials for Chemical Drugs, the Appendix for the Provision for Drug Registration. Available from: http:// www.sda.gov.cn/webportal/portal.po?UID=DWV1_WOUID_URL_53384&TOC=CO LUMN_53384&OBJ=17468518
Note: All the references can be available only in Chinese version. SDA's official website is http://www.sda.gov.cn/
Corresponding Author: Li Hui Zhen, International Medical and Commercial College, China Pharmaceutical University, NanJing, 210009, China. lihuizhenCPU@hotmail.com
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps