J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(2):223-230, 2003
HPLC determination of amoxicillin comparative bioavailability in healthy volunteers after a single dose administration.
Luis Renato Pires de Abreu1, Rodrigo Agustin Mas Ortiz
Department of Pharmacology, State University of Campinas-UNICAMP, Campinas, São Paulo, BrazilSilvana Calafatti de Castro, José Pedrazzoli Jr.
Integrated Unit of Pharmacology and Gastroenterology-São Francisco University Medical School, Bragança Paulista, São Paulo, Brazil.Received 10 July 2002, Revised 28 April 2003, Accepted 16 June 2003
PDF version
Abstract
PURPOSE: An accurate, precise and sensitive HPLC assay was developed for the determination of amoxicillin in human plasma samples, to compare the bioavailability of two amoxicillin capsule (500mg) formulations (Amoxicilina from Brazil, as a test formulation and Amoxil® from SmithKline Beecham Laboratories Ltda., Brazil, as a reference formulations) in 24 volunteers of both sexes. METHODS: Amoxicillin concentrations were analyzed by combined reversed phase liquid chromatography and UV detection (λ=229 nm). Amoxicillin and cefadroxil (internal standard) were extracted from the plasma by addition of cold methanol. The separation was achieved using the Lichrosorb® 10mm, C18 reversed phase column at room temperature. The mobile phase consisted of a 95% phosphate buffer (0.01mol/L), pH=4.8 and 5% acetonitrile mixture. The study was conducted using an open randomized 2-period crossover balanced design with a 1-week washout period between the doses. Plasma samples were obtained over an 8-hour period. The bioequivalence between the two formulations was assessed by calculating individual peak plasma concentrations (Cmax ) and area under the curve (AUC0-8h ) ratios (test/reference). The statistical interval proposed was 80-125%, as established by the US Food and drug administration Agency. RESULTS: The internal standard and amoxicillin eluted about 4.2 and 5.2 min, respectively at a flow rate of 1.3ml/min. The mean absolute recovery of AMO in plasma was 90.0% at 3mg/ml, 98.6% at 25mg/ml and 95.3 at 50mg/ml. The assay showed excellent relationships between peak height ratios and plasma concentrations (r2≥ 0.999). The limit of quantification was 1g/ml, based on 200l of plasma. The geometric mean of Amoxicilina/Amoxil® 500mg capsules individual percentage ratio was 101.4% for AUC0-8h, and 99.9% for Cmax. The 90% confidence intervals were 98.3-104.4% and 95.7-103.9%, respectively. CONCLUSION: This simple, rapid and selective method is suitable for pharmacokinetic, bioavailability and bioequivalence studies. Since the 90% CI for both Cmax and AUC0-8h lies within the 80-125% interval proposed by the Food and Drug Administration, it was concluded that Amoxicilina 500mg capsules was bioequivalent to Amoxil® capsules 500mg, in terms of both the rate and extent of absorption.
Introduction
Amoxicillin [[2S-[2a,5a,6β(S*)]]-6-[[Amino(4-hydroxyphenyl)acetyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxilic acid] is an oral semi-synthetic penicillin structurally related to ampicillin (Figure 1). The presence of a benzyl ring in the side chain extends the antibacterial activity to gram-negative bacteria (1-4). The action mechanism of these antibiotics has not been unequivocally established, but it is thought they may interfere with peptidoglycan bacterial cell wall synthesis in the effected organisms (5-10).
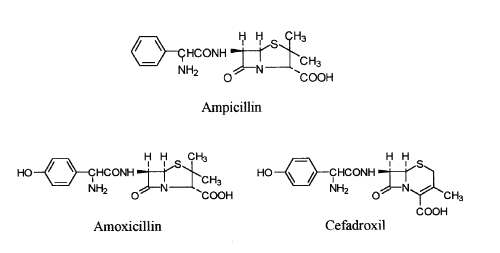
Figure 1: Structures of ampicillin, amoxicillin and cefadroxil (internal standard).
Amoxicillin (AMO) shows high absorption after oral administration, and this is not altered by the concomitant ingestion with food (6-8). AMO reaches Cmax (8mg/ml) about 2 hours after administration, exhibits low binding with plasma proteins (17%), is quickly distributed through the body, and has an elimination half-life of 1 hour (7-10). The elimination of the drug occurs preferentially by excretion in the urine, with about 60% of an orally administered dose and 75% of a parenteral dose being excreted unchanged (11-13).
AMO is commercially available in the form of capsules and tablets containing 250 and 500mg (as amoxicillin free base) for oral administration. It is also available in the form of suspensions containing 125 and 250mg/5ml.
AMO is presently the most commonly used antibiotic.To understand the pharmacokinetic behavior of AMO in humans, a reliable quantitative method is needed. Several high performance liquid chromatography (HPLC) methods for the determination of amoxicillin in body fluids have been developed. Most of the methods use direct UV detection at low wavelengths (λ=225-330 nm) (14-19). Fluorimetric detection (20-22), ion-paired reagents and special techniques such as postcolumn derivatization or column switching (14, 20, 23) have been used to enhance sensitivity and selectivity. Different methods of sample preparation have been applied prior to the chromatographic analysis, mostly based on protein precipitation (15, 19, 20), liquid-liquid extraction (21) or more complicated extractions such as solid-phase extraction (17, 18, 24-26).
The objective of this study was to compare the pharmacokinetic profiles and to evaluate the bioequivalence of two AMO formulations, in 24 healthy volunteers of both sexes. The test AMO capsule (500mg) formulation from Brazil was compared with a commercial AMO capsule (500mg) formulation produced by SmithKline Beecham, which has been defined as the reference standard by the national regulatory agency (ANVISA). After evaluation of the various conditions of the HPLC assay, a suitable and simple assay for the measurement of amoxicillin in human plasma was developed using reversed-phase HPLC and direct UV detection.
Subjects, material and methods
Drugs and chemicals
Acetonitrile (ACN) was purchased from EM Science (Gibbstown, NJ, USA); methanol (MeOH), sodium hydroxide (NaOH) and sodium phosphate salts (Na2 HPO4 /NaH 2 PO 4 ) were purchased from Merck (Rio de Janeiro, RJ, Brazil). The HPLC grade solvents ACN and MeOH were used as received. All other reagents were analytical grade. Amoxicillin and cefadroxil (internal standard) were purchased from USP Pharmacopeia (New York, NY, USA). The deionized water was prepared using Milli-Q system (Millipore, Molsheim, France).
Standard solutions
Stock solution of AMO (1mg/ml) was prepared in H2 O:ACN (95:5). Working standard solutions were prepared from the stock solution by sequential dilution with H2 O:ACN (95:5) to yield final concentrations of 1, 10 and 100 μg/ml. Stock solution of cefadroxil (internal standard) was prepared in MeOH (1mg/ml). Stock and working standard solutions were protected from light and stored at -20°C until used. Calibration standards were obtained by adding known amounts of AMO to drug-free plasma to achieve the concentrations of 1, 5, 10, 20, 40 and 50mg/ml. Three quality controls of low (3μg/ml), middle (25mg/ml) and high (50mg/ml) concentrations were prepared by adding known amounts of AMO to drug-free plasma. Plasma solutions were protected from light and stored at -70°C until used.
Extraction procedure
AMO and cefadroxil (internal standard) were extracted from human plasma samples by protein precipitation. A 200m aliquot of each plasma sample was transferred to a 1.5 ml polypropylene tube. Then, 15ml of 1 mg/ml internal standard solution and 400ml of cold MeOH (kept on ice) were added. After a brief vortex mixing, the tubes were centrifuged (14000 rpm at 4°C for 15 min). A 100m aliquot of the supernatant was transferred to the injection vials and 20ml were injected into the chromatographic system. Quality controls were performed in duplicate for each batch. All samples from a single volunteer were analyzed on the same day in order to avoid inter-assay variation.
Instruments and chromatographic conditions
The analyses were performed on a Shimadzu chromatographic system (Kyoto, Japan) equipped with a LC-10AD VP pump, an SIL-10A VP auto-sampler, an SPD-10A VP UV detector and an SCL 10 A-VP controller unit. The drug analysis data were acquired and processed using CLASS-VP (v.6.2) software running under Windows 98 on a Pentium PC. The mobile phase involved a mixture of phosphate buffer (0.01mol/L), pH=4.8 and ACN (95:5 v/v), pumped at a flow rate of 1.3ml/min through the column (Lichrosorb® 10mm RP 18, 250 x 4.6mm with a guard column Securityguard® C18 10mm, 4 x 3.0mm; Phenomenex, CA, USA) at room temperature. Peaks were monitored by UV absorbance at 229nm, sensitivity of 0.005AUFS. Quantification of AMO was obtained by plotting AMO to internal standard peak height ratios as a function of concentration.
Stability
Drug-free plasma was spiked with known amounts of the drug to achieve the concentrations of 3, 25 and 50mg/ml (n=3) and stored at -70°C. Those samples were used to investigate the stability of AMO over a period of 1 month. No internal standard was added prior to the analysis.
Specificity
The specificity of the method was determined by comparing the chromatograms obtained from the samples containing AMO and internal standard with those obtained from blank samples.
Limit of detection (LOD) and limit of quantification (LOQ)
LOD is a parameter that provides the lowest concentration of analyte in a sample that can be detected, but not quantified, under the stated experimental conditions. The LOD was determined by using the signal-to-noise ratio and comparing test results from samples with known concentrations of analyte against blank samples. The analyte concentration that produced a signal-to-noise ratio of 3:1 was accepted as the LOD. The LOQ is defined as the lowest concentration of analyte that can be determined with acceptable precision and accuracy under the stated experimental conditions. The LOQ was estimated by analyzing samples with known amounts of AMO, at progressively lower concentrations, starting at the lower end of the calibration curves. The LOQ was considered as the concentration level in which accuracy and precision were still better than 20%.
Recovery and linearity
The analytical recovery of AMO was determined at concentrations of 1, 3, 25 and 50mg/ml (n=3). Drug-free plasma was spiked with known amounts of the drug to achieve the concentration previously specified. These samples were processed by the analytical method described above and peak heights were compared with the peak heights obtained by direct injection of the drugs in the mobile phase. The linearity study was carried out in the range of 1 to 50mg/ml (n=4). To access linearity, drug-free plasma was spiked with known amounts of the drug to achieve the concentrations of 1, 5, 10, 20, 40 and 50mg/ml.
Precision and accuracy
Precision was determined as the coefficient of variation (CV), and the accuracy as the percentage relative error (RE). Precision and accuracy data were obtained by analyzing aliquots of three spiked plasma at low (3mg/ml) middle (25mg/ml) and high (50mg/ml) concentration levels of AMO. Intra-day reproducibility was determined by analyzing 5 aliquots of spiked human plasma and inter-day reproducibility was determined over a 5-day period (n=5).
Formulations
The following test formulation was employed: 500mg Amoxicilina capsules from Brazil (AC 309/Production date 03/2000; Expiry date 03/2002). The details of the reference formulation are as follows: 500mg Amoxil® capsules produced by SmithKline Beecham (BB0028/Production date 01/2000; Expiry date 01/2002).
Clinical protocol
Twenty-four (12 male and 12 female) adult volunteers, non-smokers, aged between 21 and 47 years, weighing between 55 and 95 kg and within 15% of the ideal body weight, were selected for the study. The volunteers were not on concomitant medications and were free from significant cardiac, hepatic, renal, pulmonary, gastrointestinal, neurological or hematological disease as determined within four weeks prior to the beginning of the study by way of medical histories, physical examinations and the following laboratory screenings: fasting blood glucose, urea, creatinine, SGOT (AST), SGPT (ALT), total bilirubin, total protein, plasma albumin, alkaline phosphatasis, sodium, potassium, chloride, uric acid, urinalysis, hemoglobin, hematocrit and total differential white blood cells count. All volunteers gave written informed consent and the Ethics Committee of São Francisco University Hospital approved the clinical protocol.
The study had an open randomized two-period crossover design with a 7-day washout period between doses. During each period, volunteers were hospitalized, had a regular meal, and received a 500mg capsule of the AMO allocated according to the appropriate dose randomization code. After administration of the capsule, the volunteers were asked to drink 200ml of tap water. Blood samples for plasma drug assay were taken from a forearm vein at 0, 0.5, 1, 1.5, 2, 3, 4, 6 and 8 hours after AMO administration. On each occasion, one 5ml sample was taken via "butterfly" into a clean tube. After blood clotting at room temperature, the blood samples were centrifuged at 2000g for ten minutes and the plasma removed and stored at -70°C until assayed. The volunteers received 200ml of tap water drink three hours after dose administration. Six hours after dose administration a standard lunch was made available and an evening meal was provided twelve hours after administration of the dose. Liquids were allowed ad libitum after lunch, but xanthine-containing beverages, such as tea, coffee and cola were not permitted.
Pharmacokinetic and statistical analysis
Maximum observed plasma concentration (Cmax ) and time taken to reach it (Tmax ) were obtained from drug concentration vs. time curves. The areas under the AMO concentrations vs. time curves from 0-8 hours (AUC0-8h ) were calculated using the trapezoidal method and the first order elimination rate constant (Ke) was estimated using the least square regression of the points describing the terminal log-linear decaying phase. T1/2 were derived from Ke (T1/2 = ln2/Ke). Cmax and AUC0-8h data were analyzed statistically using both parametric (one-way ANOVA) and non-parametric methods (Wilcoxon's signed ranks test).
Results
The alternative HPLC-UV method described and used here for drug quantification provides the appropriated sensitivity, specificity and high sample throughput required for pharmacokinetic studies. Figure 2 shows that under described chromatographic conditions, the retention times for AMO and internal standard were 4.2 and 5.2min, respectively. As also shown in Figure 2, no endogenous interfering peaks appeared at the retention times of the compounds of interest.
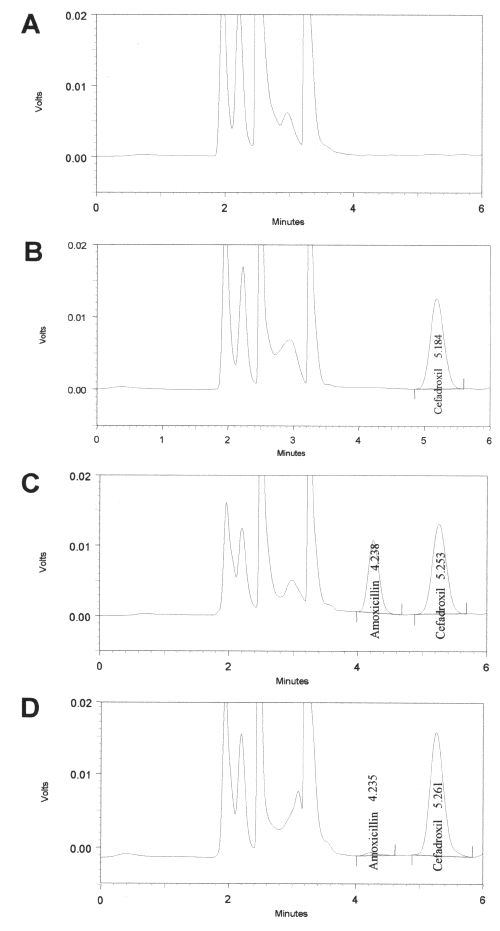
Figure 2: Chromatographic analysis of amoxicillin A) plasma blank; B) plasma + internal standard (cefadroxil 15mg/ml); C) plasma + amoxicillin (20mg/ml) + internal standard (cefadroxil 15mg/ml); D) Chromatogram obtained from plasma of a volunteer following oral administration of Amoxicillin (500mg).
The mean absolute recovery of AMO in plasma was 90.0% at 3μg/ml, 98.6% at 25mg/ml and 95.3 at 50mg/ml. The LOD and the LOQ for AMO were 0.1 and 1g/ml, respectively. The calibration curve was linear over the range 1.0g/ml to 50g/ml, with a regression coefficient ≥ 0.999 and intercept not significantly different from zero (Figure 3).
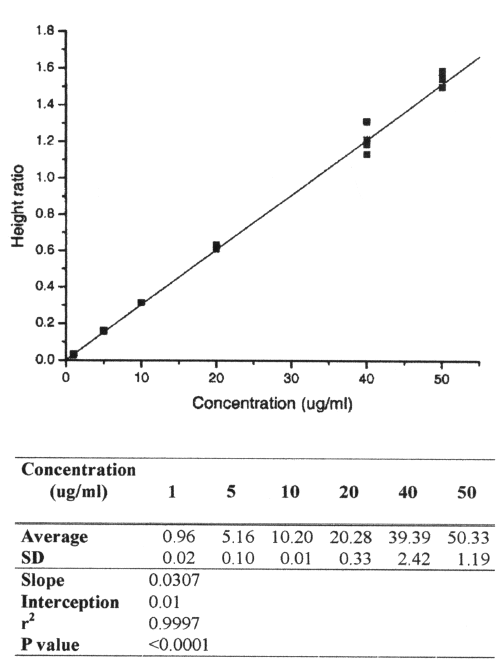
Figure 3: Calibration curve of amoxicillin.
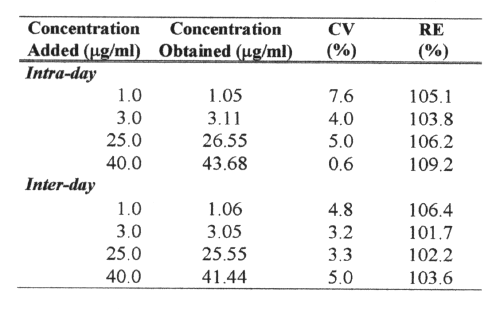
The analytical precision and accuracy obtained for intra-day and inter-day assays of four quality controls (1, 3, 25 and 50mg/ml; n=5) are shown in Table 1.
Table 1: Analytical precision and accuracy of the determination of amoxicillin from spiked plasma samples (n=5).
The overall variability (n=48) was 11.0, 6.5, 5.2 and 4.8% respectively; and the accuracy was 102.8, 103.3, 103.0, 106.8%. No significant degradation of AMO was observed during this period under the storage conditions.
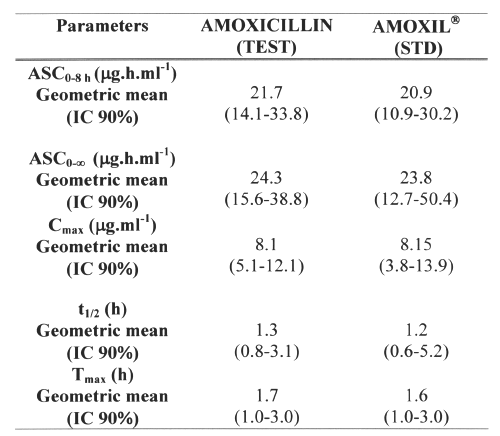
AMO was well tolerated at the administered dose, and no adverse effects were reported. Figure 4 shows mean AMO plasma concentrations as a function of time after the oral administration of 500mg AMO of both brands. The major mean pharmacokinetic parameters derived from the plasma concentration vs. time curves are presented in Table 2. The geometric mean ratios for AUC0-8h and Cmax of the two AMO oral formulations are shown in Table 3, along with analyses of their position in relation to the 80-125% interval using different tests.
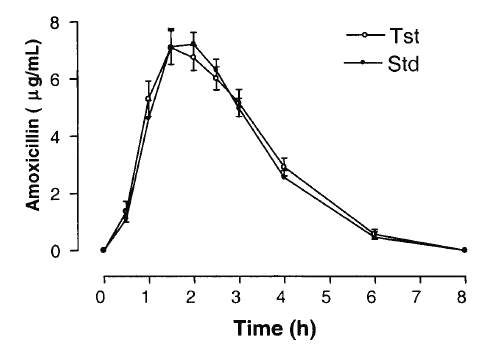
Figure 4: Curves of the mean plasma concentration of amoxicillin (± SEM) of 24 volunteers vs. time (h).
Table 2: Mean pharmacokinetic parameters obtained in 24 healthy volunteers after the administration of both 500mg amoxicillin formulations.
Table 3: Statistical analysis of AUC 0-8h and C max ratios between Amoxicilina (Test) and Amoxil® (Standard).
Discussion
AMO plasma levels have been measured by other methods using HPLC in combination with UV detection (14-19). The lowest limit of quantification obtained by UV detection was 50ng/ml, but the elution times for both AMO and internal standard were 31.8 and 32.8 min, respectively, much longer than those reported herein (14). The method of Chulavatnatol & Charles was described only for the measurement of amoxicillin in urine (19). A simple high-performance liquid chromatographic method involving protein precipitation and ultraviolet detection at 220 nm was reported by Hoizey et al. for the simultaneous determination of AMO and clavulanic acid in human plasma (15). Unfortunately, the LOQ obtained for AMO was 635mg/ml, even higher than in our method (1mg/ml). Other more complicated extraction procedures such as solid-phase extraction have also been reported (17, 18, 24-26). Nevertheless, solid-phase extraction procedures are laborious and require SPE cartridges, increasing the cost of the analysis. In order to improve sensitivity, a column switching ion-pair reversed phase liquid chromatographic system with a post column derivatization method has been used by Carlqvist & Westerlund (23). However, their assay, where the limit of quantification was 10ng/ml, has been carried out with no internal standard. In addition the method was complex due to the post column derivatization step. Their retention time was also longer for the analyte (approximately 10 min as compared to our 4 min) and additionally was not used for a pharmacokinetic study in humans where a large number of samples were analyzed.
Derivatization of AMO to a fluorescent compound showed similar sensitivity and slightly longer retention time (4.5 min for AMO), but does involve a complex chromatography system including electrochemical detection (20). The assays published by Miyazaki et al. and Mascher & Kikuta have satisfactory limits of quantification but do not utilize an internal standard, which is required for an HPLC method meeting GLP guidelines (20, 22). In a recent study, Oliveira et al. reported the determination of AMO using LC-MS-MS detection. Mass spectrometers, however, are expensive and not readily available. Furthermore, the solid-phase extraction procedure employed need laborious sample pre-treatment and requires SPE cartridges, increasing the cost of the analysis (26).
The method presented here is less sensitive than many other available assays, i.e., 1 mg/ml. However, this level of sensitivity was sufficient to perform the bioequivalence study. The pharmacokinetic study was done in 8 hours and the resulting value of AUC0-¥∞ was less than 20% higher than that of AUC0-8 for both formulations (12 and 13.9% for Amoxicilina and Amoxil respectively), as recommended by the regulatory agency. Later samples (12 and 24h after the administration of the drug) were taken and analyzed, but no better estimation of AUC0-¥∞ was observed and the differences between the values remain virtually the same. The choice of Cefadroxil as an internal standard for AMO was based on the presence of similar functional groups in both structures in addition to their similarity in terms of elemental compositions and chemical behavior. Several wavelengths (225-330nm) were evaluated and 229 nm produced the best results in terms of selectivity and sensitivity. Considering the fact that the present method involves a shorter running time and a simple sample preparation process, it may be used in similar studies as a time and cost effective alternative to other available methods.
Conclusions
The validated HPLC method employed here proved to be simple, fast, reliable, selective and sensitive enough to be used in clinical pharmacokinetic studies of AMO in humans. The median Tmax and Cmax values for both oral formulations were similar to those reported in the literature. The present study shows that Amoxicilina 500mg capsules was bioequivalent to Amoxil® capsules 500mg, in terms of both the rate and extent of absorption, as assessed by the appearance of 90% CI for both AUC0-8h and Cmax ratios within the 80-125% interval proposed by the Food and Drug Administration .
List of Abbreviations
- ACN - Acetonitrile
- AMO - Amoxicillin
- ANVISA - Agência Nacional de Vigilância Sanitária
- AUC - Area Under the Plasma Concentration-Time Curve
- CI - Confidence Interval
- Cmax - Maximal Observed Plasma Concentration
- CV - Coefficient of variation
- HPLC - High-Performance Liquid Chromatography
- Ke - Elimination rate constant
- LC-MS-MS - Liquid Chromatography-Tandem Mass spectrometry
- LOD - Limit of detection
- LOQ - Limit of quantification
- M - Mean of Experimentally determined concentration
- RE - Relative Error
- SD - Standard deviation
- T - Theoretical concentration
- T1/2 - Half-life
- Tmax - Time taken to reach Cmax
- UV - Ultraviolet
References
Tomas, A., From penicillin-binding proteins to the lysis and death of bacteria. A 1979 review. Rev Infect Dis,1: 434, 1979.
Tomas, A., The mechanism of irreversible antimicrobial effect of penicillins: how the beta lactam antibiotics kill and lyse bacteria. Ann Rev Microbiol, 33: 113,1979.
Blumberg, P. M., and Strominger, J. L., Interaction of penicillin with the bacterial cell: penicillin-binding proteins and penicillin-sensitive enzymes. Bacterial Rev, 38: 291, 1974.
Waxman, D. J., and Strominger J. L., Penicillin-binding proteins and the mechanisms of action of beta-lactam antibiotics. Annu Rev Biochem, 52: 825, 1983.
Sutherland, R., and Rolinson, G. N., Amino-p-hydroxybenzil penicillin (BRL 2333), a new semi synthetic penicillin: in vitro evaluation. Antimicr Ag Chemother, 411, 1970.
Neu, H. C. and Winshell, E. B., Pharamcological studies of 6 [D(-)-amino-p-hydroxyphenylacetamidol] penicillanic acid in humans. Antimicr Ag Chemother, 423-26, 1970.
Gordon, R. C.; Gamey, C.; Kirby, W. M. M., Comparative clinical pharmacology of amoxicillin and ampicillin administered orally. Antimicr Ag Chemother, 1: 504-07, 1972.
Kosmidis, J.; Willians, D.; Andrews, J., Amoxicillin - pharmacology, bacteriology and clinical studies. Br J Clin Pract, 26: 341-46, 1972.
Handsfield, H. H.; Clark, H.; Wallace, J. F.; Holmes, K. K.;Turck, M., Amoxicillin, a new penicillin antibiotic. Antimicr Ag Chemother, 3: 262-265,1973.
Neu, H. C., Antimicrobial activity and human pharmacology of amoxicillin. J Infect Dis, 129 (suppl): S123-S131, 1974.
Paintaud, G; Alván, G.; Dahl, M. L.; Grahnén, A.; Sjövall, J.; Svenson, J. O., Nonlinearity of amoxicillin absorption kinetics in human. Eur J Clin Pharmacol, 43: 283-288, 1992.
Simon, C.; Stille, W., Antibiotikatherapie in Klinik und Praxis. Schattauer, NY, USA,1985.
Barr, W.; Zola, E. M.; Candler, E.L., Differential absorption of amoxicillin from the human small and large intestine. Clin Pharmacol Ther, 56: 279-285, 1994.
Muth, P.; Metz, R.; Beck, H.; Bolten, W. W.; Vergin, H., Improved high-performance liquid chromatographic determination of amoxicillin in human plasma by means of column switching. J Chromatogr A, 729: 259-266, 1996.
Hoizey, G.; Lamiable, D.; Frances, C.; Trenque, T.; Kaltenbach, M.; Denis, J.; Millart, H., Simultaneous determination of amoxicillin and clavulanic acid in human plasma by HPLC with UV detection. J Pharm Biomed Anal, 30:661-6, 2002.
Sourgens, H.; Steinbrede, H.; Verschoor, J. S.; Bertola, M. A.; Rayer, B., Bioequivalence study of a novel Solutab tablet formulation of amoxicillin/clavulanic acid versus the originator film-coated tablet. Int J Clin Pharmacol Ther, 39:75-82, 2001.
Yuan, Z.; Russlie, H. Q.; Canafax, D. M., Sensitive assay for measuring amoxicillin in human plasma and middle ear fluid using solid-phase extraction and reversed-phase high-performance liquid chromatography. J Chromatogr B, 674:93-9, 1995.
Krauwinkel, W. J.; Volkers-Kamermans, N. J.; van Zijtveld, J., Determination of amoxicillin in human plasma by high-performance liquid chromatography and solid phase extraction. J Chromatogr, 617:334-8, 1993.
Charles, B. and Chulavatnatol, S., Simple analysis of amoxicillin in plasma by high performance liquid chromatography with internal standardization and ultraviolet detection. Biomed Chromatogr, 7:204-7, 1993.
Marscher, H.; Kikuta, C., Determination of amoxicillin in plasma by high-performance liquid chromatography with fluorescence after on-line oxidation. J Chromatogr A, 506: 417-421, 1990.
Wibawa, J. I.; Fowkes, D.; Shaw, P. N.; Barrett, D. A., Measurement of amoxicillin in plasma and gastric samples using high-performance liquid chromatography with fluorimetric detection. J Chromatogr B Analyt Technol Biomed Life Sci, 774:141-8, 2002.
Miyazaki K, Ohtani K, Sunada K, Arita T. Determination of ampicillin, amoxicillin, cephalexin, and cephradine in plasma by high-performance liquid chromatography using fluorometric detection. J Chromatogr, 276:478-82, 1983.
Carlqvist, J. and Westerlund, D., Automated determination of amoxicillin in biological fluids by column switching in ion-pair reversed phase liquid chromatographic systems with post-column derivation. J Chromatogr, 344: 285-296, 1985.
Maurer, H. H., Liquid chromatography-mass spectrometry in forensic and clinical toxicology. J Chromatogr B, 713: 3-25, 1998.
Henion, J.; Brewer, E.; Rule, G., Sample preparation for LC/ MS/MS: analyzing biological and enviromental samples. Anal Chem, 70:650-656, 1998.
Oliveira, C. H.; Abib, E.; Vannuchi, Y. B.; Sucupira, M.; Ilha, J.; De Nucci, G., Comparative bioavailability of 4 amoxicillin formulations in healthy human volunteers after a single dose administration. Int J Clin Pharm And Ther, v.39, 4: 167-172, 2001.
Corresponding Author: Luis Renato Pires de Abreu, 288, Rua Simão Ferreira Alves. CEP: 13801-525, Mogi Mirim, SP-Brazil. lrpabreu@uol.com.br
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps