J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(2):189-197, 2003
Poloxamer 407-mediated alterations in the activities of enzymes regulating lipid metabolism in rats.
Kishor M. Wasan1, Ramaswamy Subramanian, Mona Kwong
Division of Pharmaceutics and Biopharmaceutics, Faculty of Pharmaceutical Sciences, University of British Columbia, Vancouver, British Columbia, CanadaIra J. Goldberg, Thamrah Wright
Department of Preventive Medicine and Nutrition, Columbia University College of Physicians and Surgeons, New York, New York, USAThomas P. Johnston
Division of Pharmaceutical Sciences, School of Pharmacy, University of Missouri, Kansas City, Missouri, USAReceived 6 January 2003, Revised 10 April 2003, Accepted 22 April 2003
PDF version
Abstract
PURPOSE: Recently, the P-407-treated mouse was established as a useful animal model of hyperlipidemia and atherosclerosis. The present study was aimed to determine whether P-407-induced hyperlipidemia in the rat is associated with alterations in the activities of enzymes responsible for lipid metabolism. Methods and Results: Rats were made hyperlipidemic by i.p. injection of 1.0 g/kg P-407 and blood samples collected 24 h after administration of P-407. Plasma from P-407-treated rats demonstrated 7- and 13-fold increases in cholesterol and triglycerides, respectively (p < 0.001). The plasma lecithin cholesterol acyl transferase (LCAT) activity in these animals was 4-5-fold greater than control animals (p < 0.05). Further, the plasma cholesteryl ester transfer protein (CETP) activity in P-407-treated rats was increased by ~25%, which was inhibited by > 50% in the presence of TP2, a monoclonal anti-CETP antibody (27.03 ± 3.16 vs. 10.87 ± 3.23; p < 0.05). The plasma CETP protein levels were also increased by 5-6-fold in P-407-treated animals (control 0.35 ± 0.17 vs. P-407 treated 1.87 ± 0.35 ug/ml, p < 0.05). However, the plasma hepatic lipase (HL) (control 49.2 ± 3.1 vs. P-407-treated 2.0 ± 0.38 umol/ml/h; p < 0.001) and lipoprotein lipase (LPL) (control 45.9 +/- 0.09 vs. P-407-treated 2.03 ± 0.38 mol/ml/hr; p<0.001) activities in these animals were significantly inhibited. Conclusions: In summary, P-407-induced hyperlipidemia in rats is associated with alterations in plasma LCAT, CETP, HL and LPL activities.
Introduction
In recent years, there has been a gradual increase in mortality due to coronary heart disease (CHD) in North America (1-3). Traditionally, factors such as hypercholesterolemia, cigarette smoking, diabetes mellitus and sedentary life style have been implicated in the development of hyperlipidemia and atherosclerotic cardiovascular disease (2). However, it is now becoming increasingly evident that certain chemicals, such as surface-active agents (detergents) also have the potential to cause hyperlipidemia, should they be consumed over an extended period. One such example, Poloxamer 407 (Pluronic® F-127, P-407), has been shown to cause significant elevations in plasma cholesterol and triglycerides in various animal models, including rats (4-6), mice (7-10) and rabbits (11). In rats and mice, the elevation in plasma triglycerides appears to be more sensitive to the effects of P-407 when compared to plasma cholesterol (4). However, to date the mechanisms by which P-407 modifies plasma lipid concentrations remains unknown.
P-407 is a hydrophilic non-ionic surface-active agent with a low degree of toxicity and an LD 50 greater than 15g/kg and 1.8 g/kg following intraperitoneal (i.p.) administration in rabbits and mice, respectively (6, 11-12). P-407 has been used as an excipient for sustained delivery of water-soluble drugs delivered extravascularly (12-14). In addition to its effects on plasma cholesterol and triglycerides, P-407 has been demonstrated to cause significant inhibition of plasma lipoprotein lipase (LPL), both in vitro and in vivo accompanied by an increase in tissue LPL activity (5). The accumulation of plasma triglycerides following the administration of P-407 has, therefore, been suggested to be due to inhibition of capillary-bound LPL activity. Other studies have indicated that, in rats, the increase in plasma cholesterol following P-407 i.p. administration may be due to inhibition of cholesterol 7a -hydroxylase, but not stimulation of 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase (10). Histological evaluation of aortic valves from C57BL/6 mice exposed to P-407 showed progressively increasing atherosclerotic lesion size following 16-20 weeks of exposure (7-9). Taken together, these observations suggest that chronic exposure to P-407 result in hyperlipidemia, which may lead to atheroma formation in animal models that are traditionally regarded as difficult to induce hyperlipidemia and atherosclerosis.
A number of enzymes are responsible for the metabolism and subsequent removal of plasma cholesterol and triglycerides for the systemic circulation (15, 19, 20, 21). Lecithin cholesterol acyl transferase (LCAT) is an enzyme responsible for the conversion of cholesterol-to-cholesterol esters on the surface of high-density lipoproteins (HDL) (16). Plasma LCAT secreted by liver hepatocytes is a 63 KDa protein containing 416 amino acids and four N-linked glycosylation sites, which catalyzes the transfer of fatty acids from the sn -2 position of lecithin to the free hydroxyl group of cholesterol to form cholesteryl ester and lysophospholipid (17). Cholesteryl esters formed because of LCAT-mediated cholesterol esterification, triglycerides and to a lesser extent, phospholipids are redistributed between plasma lipoproteins by cholesteryl ester transfer protein (CETP), a 74 KDa hydrophobic glycoprotein containing 476 amino acids (22-23). CETP (24-30) is primarily secreted from the liver and adipose tissue and circulates in the plasma, bound mainly to HDL (24).
Triglycerides are hydrolyzed by lipoprotein lipase (LPL), an enzyme found in two distinct pools in the tissue (31). The fraction responsible for the hydrolysis of plasma lipoprotein triglycerides is localized on the surface of endothelial cells and is readily released upon perfusion with heparin. A second pool of LPL remains in the tissue after perfusion and is thought to represent a precursor or intracellular storage pool for the endothelium-bound enzyme. Measurement of capillary-bound enzyme activity provides an estimate of the ability of LPL to clear circulating triglycerides. Measurement of heparin-releasable LPL is the best indicator of the amount of capillary-bound enzyme (5, 32). Hepatic lipase (HL) (35-44) is another enzyme involved in the metabolism of triglycerides. Specifically, HL hydrolyzes phospholipids and triglycerides in all lipoprotein classes (33-34).
Since each of these enzymes plays a vital role in the metabolism and subsequent removal of plasma cholesterol and triglyceride the objective of the study was to determine whether P-407-induced hyperlipidemia is associated with alterations in plasma LCAT, CETP, HL and LPL activity in rats. We hypothesized that P-407 induced hyperlipidemia may be due to P-407's stimulatory effect on LCAT and CETP activity resulting in evaluated plasma cholesterol levels and P-407's inhibitory effect on LPL and HL resulting in evaluated plasma triglyceride levels.
Materials and Methods
Materials
Twenty, male Sprague-Dawley rats (225-250 g) were obtained from Charles River Inc. (Wilmington, MA). Ten of the animals were used for harvesting blood 24 hours after receiving an i.p injection of Poloxamer 407 (Pluronic® F-127; BASF Corporation; Mount Olive, NJ, USA), six were purchased with a cannula having already been placed in the external jugular vein and were used to collect post-heparin plasma for HL activity determinations, and four rats were used as controls and blood collected 24 hours after an i.p. injection of normal saline. All animals had free access to food and water and were maintained in a temperature-controlled room (22°C) on a 12:12 h light:dark cycle. Assay kits for the measurement of total and free plasma cholesterol and triglycerides were obtained from Sigma (St. Louis, MO, U.S.A). Plasma protein concentration was measured according to the Bradford method using a BioRad protein assay kit (Bio-Rad Laboratories, Hercules, CA). P-407 (Pluronic® F-127; BASF Corporation; Parsipanny, NJ, U.S.A) solution for intraperitoneal (i.p.) injection was prepared by combining the agent with sterile water for injection U.S.P., and refrigerated overnight to facilitate dissolution of P-407 by the cold method of incorporation (45). Normolipidemic fasted human plasmas (total plasma cholesterol concentrations range, 135-150 mg/dl) were obtained from Bioreclaimation Inc. (Hicksville, NY). Purified CETP was a generous gift from Dr. Richard E. Morton (Cleveland Clinic, OH). The monoclonal antibody raised up against rabbit CETP, TP2, was obtained from the Ottawa Heart Institute (Ottawa, Ontario, Canada). [3H]cholesterol (1Ci/mmol) and ([1a,2a (n)-3H] cholesteryl oleate (specific activity, 7.39mCi/mg) were purchased from Amersham Pharmacia Biotech (Oakville, Ontario, Canada). Unlabeled cholesterol, cholesteryl oleate and 5,5'-dithio-bis(2-nitrobenzoic acid) (DTNB) were obtained from Sigma Chemical (St. Louis, Mo, USA). TLC plates (20 x 20cm) were purchased from VWR (Ontario, Canada). Solvents for the development of TLC plates were purchased from Fisher Scientific (Ontario, Canada). Diethyl ether was purchased locally. All other chemicals were of reagent grade and obtained from Sigma Chemical (St. Louis, MO, USA).
Induction of hyperlipidemia
All animals were made hyperlipidemic by an i.p. injection of a 1.0 gm/kg dose of P-407. All syringes were placed on ice prior to P-407 administration to maintain the polymer in a mobile viscous state during the injection, since P-407 solutions at concentrations greater than about 23% w/w exhibit reverse thermal gelatin properties.
Collection of blood
For animals without cannulas in the external jugular vein, blood was collected from the descending abdominal aorta into heparinized (100 units/ml) glass tubes at exactly 24 hr after receiving either a dose of P-407 or normal saline. Briefly, each animal was lightly anesthetized and then the peritoneal cavity opened, the descending abdominal aorta isolated, and the blood sample (approx. 8 mL) obtained. The animal was then immediately sacrificed while under anesthesia. The blood samples were then centrifuged at 10,000 g for 10 min at 4°C, the plasma harvested, and the plasma sample frozen at -80° until the time of analysis.
Animals used for the collection of post-heparin plasma (pre-cannulated rats) were administered 2000 Units of heparin through the indwelling cannula in the left external jugular vein exactly 24 hr following i.p. administration of either P-407 or normal saline. Two minutes later, four, 1 ml blood samples were collected into 1.5 ml polypropylene microcentrifuge tubes, the tubes centrifuged at 10,000 g for 10 min at 4°C, the plasma obtained, and the samples frozen at -80°C until the time of HL activity determination. Heparin was administered to release tissue bound LPL and HL.
All procedures for P-407 administration and subsequent blood collection were in accordance with the institution's guide for the care and use of laboratory animals, and the treatment protocol was approved by the Animal Care and Use Committee at the University of Missouri-Kansas City and the University of British Columbia.
Lipid analysis
The control and P-407-treated plasma samples were analyzed for total cholesterol and triglycerides according to the procedure described in the Sigma Infinity® cholesterol and triglycerides assay kits (Sigma Chemical, St. Louis, MO, USA).
Cholesterol esterification rate
The endogenous cholesterol esterification rate was measured as a function of plasma LCAT activity as described by Dobiasova and Frohlich (46) with slight modifications. Briefly, Whatman no. 1 filter paper discs containing trace amount (0.25m Ci) of [3H]cholesterol were added to 100m l plasma and incubated overnight at 4°C to allow incorporation of the radiolabel into plasma lipoproteins without esterification. Fifty microliters of plasma containing [3H]cholesterol was incubated at 37°C for 60 min and the reaction stopped with 1ml of 95% ethanol. The precipitate formed was separated by centrifugation at 4,200 rpm for 20 min, the supernatant dried under nitrogen and the dried extract reconstituted with 50m l chloroform. Twenty microliters of the reconstituted extract was applied onto a TLC plate and the cholesterol and cholesteryl ester separated using hexane:diethyl ether:acetic acid (85:15:1). After developing in iodine, the bands corresponding to unesterified cholesterol and cholesteryl ester were scraped and the radioactivity measured by liquid scintillation counting. DNTB, a known inhibitor of LCAT, was used as a positive control in these experiments.
Separation of plasma lipoproteins
Prior to separation of lipoproteins, the plasma was incubated for 24 h at 37°C with (3H) cholesteryl oleate for incorporation of radiolabel into plasma lipoproteins. The high- and low-density lipoproteins, HDL and LDL were separated from the radiolabeled plasma by sodium bromide density gradient ultracentrifugation (47-48).
CETP activity assay
The transfer of radiolabeled cholesteryl esters between HDL and LDL was measured using delipidated rat plasma as the source of CETP according to the procedure described by Morton and Zilversmit (49,50). In these experiments (3H) CE-enriched LDL was incubated with the corresponding unlabeled HDL, along with delipidated rat plasma in a buffer containing Tris-HCl 50 mM, NaCl 150 mM, sodium azide 0.02%, disodium ETDA 0.01%, pH 7.4. In some experiments, delipidated plasma was co-incubated for 90 min at 37°C with a monoclonal antibody directed against CETP, TP2 (4m g protein) prior to studying the transfer of cholesteryl esters between lipoproteins as described above. Lipid transfer between lipoproteins was measured by liquid scintillation counting and CETP activity was calculated using the following equation
kt = -ln (1-At/Do)
where Do and At are the radioactivities of the donor at time 0 and the acceptor at time t, respectively. The constant k is the fraction of label transferred per unit time (t). Acceptor radioactivity in the absence of CETP (usually < 2-3%) was subtracted before calculating kt values. Calculations assume steady-state conditions where all lipid and drug transfer are in an exchange process (49,50).
Measurement of CETP protein
Plasma CETP protein concentration was measured by an enzyme-linked immunosorbent assay (ELISA) using an enzymatic assay kit obtained from Wako Pure Chemical Industries Ltd. (Osaka, Japan). The CETP protein concentration was expressed as m g/ml.
Lipase assay
Hepatic lipase (HL; EC 3.1.1.3) was assayed in post-heparin plasma according to a modified assay as originally described by Hocquette et al . (51). This measures total post heparin lipolytic activity. HL is specifically assessed by performing the assay in high salt (1 M NaCl). In this assay, HL activity was measured by quantifying the amount of fatty acid produced from the enzyme-catalyzed hydrolysis of an intralipid emulsion radiolabeled with [3H]triolein. The substrate emulsion contained 10% Intralipid® (120 mM of TG; Baxter Corp., Chicago, IL, USA) into which a trace amount of [H3]triolein (approximately 14MBq) had been incorporated by sonication on ice (50% pulse,100 W, 10 min) using a Braun-Sonic U Type 853973/1 sonicator. The incubation medium was prepared from 10 ul intralipid emulsion, 10 ul heat-inactivated serum as a source of apolipoprotein CII, 60 ul of de-ionzed water and 100 ul of incubation buffer which contained 12% fatty acid free bovine serum albumin, 0.2% standard heparin, 1.0 M NaCl and 0.3 Tris-HCL, pH 8.5. To initiate LPL inhibition, 5 ul of rat post-heparin plasma was incubated in 1.0 M NaCl for 5 minutes prior to adding the incubation medium. Each assay tube contained a total volume of 200 ul and a final concentration of 1 M NaCl. The samples were incubated in triplicates at 25°C for 60 minutes and the reaction stopped by adding 3.5 mL of methanol:chloroform:heptane (56:50:40 v:v:v). The liberated fatty acid was isolated from the unhydrolysed triglyceride by a selective extraction procedure in which 1 ml of borate buffer was added, the tubes vigorously vortexed and then centrifuged at 2000 rpm for 20 min at 4°C in a Beckman Coulter Allegra 6KR centrifuge. The fatty acid product was then quantitated by scintillation counting a 1.0 ml aliquot of the aqueous phase in 3.5 ml of EcoscintTM H scintillation fluid (National Diagnostics LS-275) for 1 min in a Beckmann LS 7500 scintillation counter (Fullerton, CA). Hepatic lipase activity was then calculated in terms of specific activity based on the volume of PHP as μmol FFA liberated/ml/ hour.
LPL activity was determined by subtracting HL activity from post heparin lipolytic activity. This was measured as described above but without inclusion of the high salt buffer.
Data analysis
Data is expressed as mean ± S.E.M. All the data, except LCAT activity was analyzed by student `t' test. LCAT activity data was analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test. Data was considered to be statistically significant if p < 0.05.
Results
Plasma lipids
48 hours after a single i.p. injection of P-407, the plasma from the P-407-treated rats exhibited marked hyperlipidemia, as demonstrated by a 7-8-fold increase in total cholesterol (459.1 ± 51.8 vs. 67.1 ± 3.8 mg/dl, p < 0.001) (Figure 1) and 12-13-fold increase in total plasma triglycerides (926.2 ± 73.9 vs. 72.5 ± 9 mg/dl, p < 0.001) (Figure 2) compared to saline-treated control rat plasma.
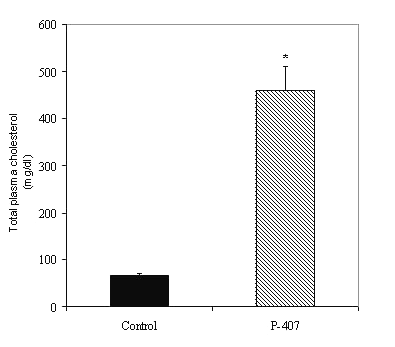
Figure 1: Plasma total cholesterol levels in saline-treated control (n=5) and P-407-treated (n=12) rats. * p < 0.001 vs. saline-treated control.
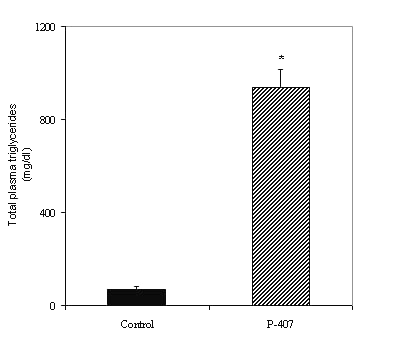
Figure 2: Triglyceride levels in plasma obtained from saline-treated control (n=4) and P-407-treated (n=9) rats. * p < 0.001 vs. saline-treated control.
Enzyme activities
Plasma obtained from P-407-treated rats also displayed elevated LCAT activity, as demonstrated by a significant increase in endogenous cholesterol esterification rate (138.3 ± 31.4 nmol/ml/h) compared to the control rat plasma (29.8 ± 11.3 nmol/ml/h; p < 0.05) (Figure 3).
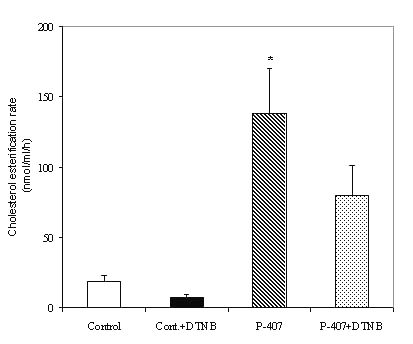
Figure 3: LCAT activity in saline-treated control (n=3) and P-407-treated (n=6) rat plasma and the effect of 1.5 mM 5,5'-dithio-bis(2-nitrobenzoic acid) (DTNB) on plasma LCAT activity. * p < 0.05 vs. saline-treated control.
Experiments were also performed to study the effects of 1.5mM DTNB, an inhibitor of plasma LCAT activity. In control rat plasma, DTNB treatment for 30 min. inhibited the LCAT activity by ∼ 60%, whereas P-407-treated rat plasma exhibited a modest 30% inhibition (Figure 3). Experiments were performed to measure CETP activity as described in the methods. When delipidated plasma was used as the source of CETP, the P-407-treated rat plasma exhibited 27% CETP activity, which was blocked by TP2 to the extent of ∼ 60% (p < 0.05) (Figure 4).
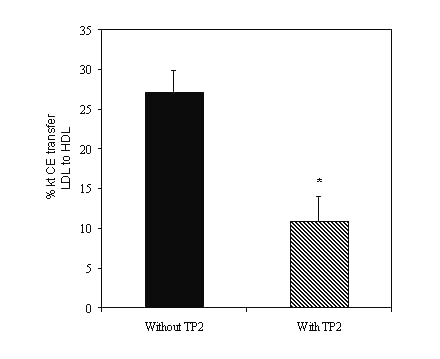
Figure 4: CETP-mediated cholesteryl ester (CE) transfer in P-407-treated rat plasma in the absence (without TP2, n=3) and presence (with TP2, n=3) of monoclonal anti-CETP antibody, TP2 (4m g). No CETP activity was detected in saline-treated control rat plasma. * p < 0.05 vs. without TP2 .
In saline-treated control rat plasma, CETP activity was not detected both in the absence or presence of TP2. The CETP activity measured in P-407-treated rat plasma was higher than the activity observed with human plasma but comparable with the rabbit CETP activity (data not shown). Additional experiments were performed to measure CETP protein levels in the rat plasma. Plasma obtained from P-407-treated rats exhibited a 6-fold increase in CETP protein levels as measured by ELISA compared to the corresponding saline-treated control rat plasma (0.35 ± 0.17 vs. 1.87 ± 0.35 m g/ml; p < 0.05) (Figure 5).
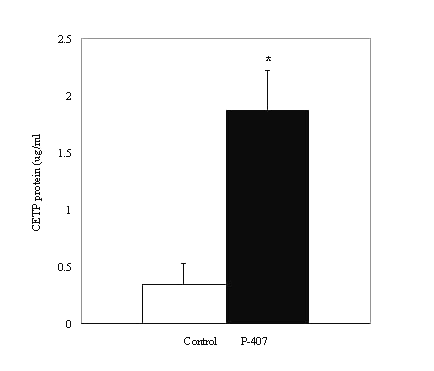
Figure 5: CETP protein levels in saline-treated control (n=3) and P-407-treated (n=6) rat plasma. * p < 0.05 vs. saline-treated control.
The activity of LPL and HL in post-heparin plasma 24 hr after an i.p. injection of P-407 was virtually negligible (1.11 ± 0.09 and 2.03 ± 0.38 m mol/ml/hr, respectively) (p<0.001) when compared to the lipase activity determined for controls [45.9 ± 13.1 (LPL) and 49.2 ± 3.1 (HL)m mol/ml/hr] (Figure 6).
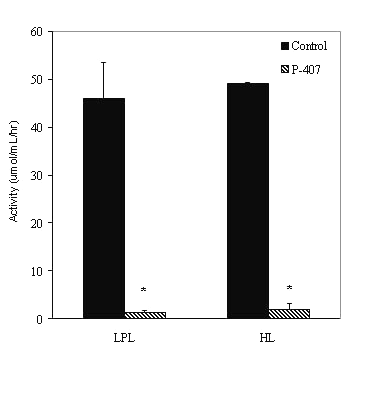
Figure 6: Lipoprotein lipase (LPL) and hepatic lipase (HL) activity in saline-treated control (n=3) and P-407-treated (n=6) rat plasma. * p < 0.001 vs. saline-treated control.
DISCUSSION
In previous studies, a single i.p. injection of P-407 produced marked hyperlipidemia in rodents (4-10). This was confirmed in the present study, where the plasma cholesterol and triglyceride levels were significantly elevated in the P-407-treated animals, compared to the saline-treated controls (Figures 1 & 2). In addition to hyperlipidemia, previous studies have also demonstrated that extravascular administration of P-407 produces atherosclerotic lesions in mice (7-9). A novel finding from the present study is that, in rats, P-407-induced hyperlipidemia is also associated with alterations in the activities of enzymes that are responsible for cholesterol transport and metabolism. Plasma LCAT and CETP activities are markedly elevated in animals that are treated with P-407, whereas, the plasma LPL and HL activities are significantly inhibited as shown in Figures 3, 4 and 6. Furthermore, the elevated plasma CETP activity appears to be due to increased plasma CETP protein concentration following P-407 administration as shown in Figure 5.
The esterification of cholesterol by LCAT facilitates the flow of unesterified cholesterol from the peripheral tissues to the plasma. Thus, LCAT activity has been traditionally regarded as anti-atherogenic. The increase in LCAT activity observed in this study could, therefore, be a protective mechanism that counteracts P-407-induced hyperlipidemia. However, it has been suggested that higher LCAT activity reflected by a higher cholesterol esterification rate is not necessarily protective or anti-atherogenic (17). It is also possible that the elevated plasma LCAT activity in the P-407-treated animals is a compensatory mechanism that accelerates the metabolism and clearance of cholesterol from the plasma due to increased esterification of cholesterol. However, observations from a recent in vitro study in ( 3 H) cholesterol-labeled fibroblasts suggest that the efficiency of cholesterol efflux in LCAT-deficient plasma is comparable with normal plasma (52). In addition to LCAT, other factors, such as increased oxidized LDL, increased pre beta HDL or altered nitric oxide levels and the generation of oxidative free radicals may be responsible for the development of atherosclerotic lesions in P-407-treated rats.
Traditionally, the rat has been regarded as an animal devoid of CETP activity (53,54). However, in a study by Jiang et al ., CETP-like mRNA was detected by RNase protection analysis in several rat tissues, namely, heart, skeletal muscle, adipose tissue and small intestine (30). An interesting observation from the present study is the detection of CETP activity and CETP protein in the P-407-treated rat plasma. A plausible explanation for this observation is that, in rats, long-term administration of P-407 causes posttranscriptional upregulation of CETP protein as well as the corresponding activity, which would facilitate increased transfer of cholesteryl esters between plasma lipoproteins.
Inhibition of HL activity by P-407 was not totally unexpected. Previously, it was shown that a different lipase, namely, the heparin-releasable fraction of LPL was significantly inhibited following a single i.p. injection of P-407 to rats compared to controls (5). P-407-mediated inhibition of HL further supports the results observed by others with regard to HL activity and atherosclerosis. Jansen et al. states that in experimental animals that normally have low plasma CETP activity (e.g., rodents), HL activity is often inversely associated with atherosclerosis (44). Our model would tend to support this, since P-407-treated mice develop extensive aortic atherosclerosis after approximately 16-20 weeks (7,8) despite having an elevated plasma HDL cholesterol concentration. Jansen et al. further suggest that plasma HDL cholesterol concentration is often a poor indicator of atherogenic risk in animal models (44). Again, this would appear to be the case with the P-407-induced mouse model of hyperlipidemia and atherosclerosis.
Systemic exposure to P-407 at low concentrations appears to modulate the activities of CETP, LCAT, LPL and HL in support of enhanced reverse cholesterol transport. Although Dietschy and Turley (56) have shown that net cholesterol flux from peripheral tissues to the liver is not dependent on the plasma concentrations either HDL-C or apoA-I, nevertheless, changes in the activities of CETP, LCAT and HL can and do affect the plasma concentrations of atheroprotective HDL-C (55-57). By increasing the activity of LCAT with P-407, cholesterol previously picked up from peripheral cells by nascent HDL would be esterified at a faster rate. The rate at which newly formed CE contained in HDL could be transferred to apoB-containing lipoproteins for subsequent elimination would also be increased due to the P-407-mediated increase in CETP activity. Lastly, inhibiting the activity of HL would stimulate an increase in the plasma concentration of atheroprotective HDL-C. Taken together, the combined effects of P-407 on the activities of LCAT, CETP, HL and LPL would seem to suggest that the P-407-treated rat is attempting to clear the plasma cholesterol burden as rapidly as possible. However, the fact that mice develop atherosclerotic lesions after 16-20 weeks would tend to support either additional pro-atherogenic mechanisms in this model or suggest an inability of the P-407-treated rat to completely and efficiently catabolize/eliminate the increased plasma cholesterol stores.
In conclusion, the results presented in this study suggest that P-407-induced hyperlipidemia in rats is also associated with marked alterations in the activities of enzymes that are responsible for lipid metabolism. These alterations may provide a mechanistic link for the development of atherosclerosis following systemic exposure to a surface active agent, such as P-407.
Acknowledgments
Funding for this study was provided by the Canadian Institutes of Health Research (Grant # MOP 48090) and an AAPS New Investigator Grant in Pharmaceutics and Pharmaceutical Technology to KMW.
REFERENCES
Miettinen TA. Cholesterol absorption inhibition: A strategy for cholesterol lowering therapy. Int J Clin Pract 2001; 55: 710-16.
Frohlich J, Lear SA. Old and new risk factors for atherosclerosis and development of treatment recommendations. Clin Exp Pharmacol Physiol 2002; 29: 838-42.
Kruth HS. Lipoprotein cholesterol and atherosclerosis. Curr Mol Med 2001; 1: 633-53.
Zjumira GM, Wout M, Pec EA, et al. Poloxamer 407-mediated changes in plasma cholesterol and triglycerides following intraperitoneal injection to rats. J Parent Sci Tech 1992; 46: 192-200.
Johnston TP, Palmer WK. Mechanism of Poloxamer 407-induced hypertriglyceridemia in the rat. Biochem Pharmacol 1993; 46: 1037-42.
Johnston TP, Palmer WK. Effect of Poloxamer 407 on the activity of microsomal 3-hydroxy-3 methylglutaryl CoA reductase in rats. J Cardiovasc Pharmacol 1997; 29: 580-85.
Palmer WK, Emeson EE, Johnston TP. Poloxamer 407-induced atherogenesis in the C57BL/6 mouse. Atherosclerosis 1998; 136: 115-23.
Johnston TP, Baker JC, Jamal AS, et al. Potential downregulation of HMG-CoA reductase after prolonged administration of P-407 in C57B/6 mice. J Cardiovasc Pharmacol 1999; 34: 831-42.
Johnston TP, Baker JC, Hall DD et al. Regression of Poloxamer-407-induced atherosclerotic lesions in C57BL/6 mice using atorvastatin. Atherosclerosis 2000; 149: 303-13.
Johnston TP, Nguyen LB, Chu WA, et al. Potency of select statin drugs in a new mouse model of hyperlipidemia and atherosclerosis. Int J Pharm 2001; 229: 75-86.
Blonder JM, Baird L, Fulfs JC et al. Dose-dependent hyperlipidemia in rabbits following administration of poloxamer 407 gel. Life Sci 1999; 65: 261-66.
Johnston TP, Miller SC. Toxicological evaluation of poloxamers for intramuscular use. J Parent Sci Technol 1985; 39: 83-8.
Pec EA, Wout Z, Johnston TP. Biological activity of urease formulated in poloxamer 407 following intraperitoneal injection in the rat. J Pharm Sci 1992; 81: 626-30.
Johnston TP, Punjabi MA, Froehlich CF. Sustained delivery of interleukin-2 from a poloxamer 407 gel matrix following intraperitoneal injection in mice. Pharm Res 1992; 9: 421-30.
Sviridov D, Nestel P. Dynamics of reverse cholesterol transport: protection against atherosclerosis. Atherosclerosis 2002; 161: 245-54.
Jonas A. Regulation of lecithin cholesterol acyl transferase activity. Progress Lipid Res. 1998; 37: 209-34.
Glomset JA. The plasma lecithins:cholesterol acyl transferase reaction. J Lipid Res 1968; 9: 155-67.
Hoeg JM, Santamarina-Fojo S, Berard AM, et al. Overexpression of lecithin:cholesterol acyltransferase in transgenic rabbits prevents diet-induced atherosclerosis. Proc Natl Acad Sci, USA 1996; 93: 1148-53.
Wilson PW, Abbott RD, Castelli WP. High-density lipoprotein cholesterol and mortality. The Framingham Heart Study. Arteriosclerosis 1988; 8:737-41.
Assmann G, Schultze H, von Eckardstein A, et al. High-density lipoprotein cholesterol as a predictor of coronary heart disease risk. The PROCAM experience and pathophysiological implications for reverse cholesterol transport. Atherosclerosis 1996; 124(S): 11-20.
Gordon DJ, Rifkind BM. High-density lipoprotein the clinical implications of recent studies. N Eng J Med 1989; 321: 1311-6.
Drayna D, Jarnagin AS, McLean J. et al. Cloning and sequencing of human cholesteryl ester transfer protein cDNA. Nature 1987; 327: 632-4.
Hesler CB, Tall AR, Swenson TL, et al. Monoclonal antibodies to the Mr 74,000 cholesteryl ester transfer protein neutralize all of the cholesteryl ester and triglyceride transfer activities in human plasma. J Biol Chem 1988; 263: 5020-
Tall AR. Plasma cholesteryl ester transfer protein. J Lipid Res 1993; 34: 1255-74.
Ha YC, Barter PJ. Differences in plasma cholesteryl ester transfer activity in sixteen vertebrate species. Comp Biochem Physiol 1982;71: 265-9.
Barter P. CETP and atherosclerosis. Arterioscler Thromb Vasc Biol 2000; 20: 2029-31.
Zhong S, Sharp DS, Grove JS, et al. Increased coronary heart disease in Japanese-American men with mutations in the cholesteryl transfer protein gene despite increased HDL levels. J Clin Invest 1996; 97: 2917-23.
Marotti KR, Castle CK, Boyle TP, et al. Severe atherosclerosis in transgenic mice expressing simian cholesteryl ester transfer protein. Nature 1993; 364: 73-5.
Rittershaus C, Miller DP, Thomas LJ, et al. Vaccine-induced antibodies inhibit CETP activity in vivo and reduce aortic lesions in a rabbit model of atherosclerosis. Arterioscler Thromb Vasc Biol 2000; 20: 2106-12.
Jiang X, Moulin P, Quinet E, et al. Mammalian adipose tissue and muscle are major sources of lipid transfer protein mRNA. J Biol Chem 1991; 266: 4631-9.
Oscai LB, Palmer WK. Cellular control of triacylglycerol metabolism. In: Terjung RL, ed. Exercise and Sports Sciences Reviews, Philadelphia, PA: Franklin Institute, 1983:1-23.
Borensztajn J. Lipoprotein lipase. In: Scanu AM, Wissler RH, Getz GS, eds. The Biochemistry of atherosclerosis. New York: Marcel Dekker, 1979:231-45.
Cohen J, Vega GL, Grundy SM. Hepatic lipase: new insights from genetic and metabolic studies. Curr Opin Lipidol 1999; 10: 259-68.
Thuren T. Hepatic lipase and HDL metabolism. Curr. Opin. Lipidol. 2000; 11: 277-84.
Conelly PW, Hegele RA. Hepatic lipase deficiency. Crit Rev Clin Lab Sci 1998; 35: 547-72.
Santamarina-Fojo S, Haudeschild C, Amar M. The role of hepatic lipase in lipoprotein metabolism and atherosclerosis. Curr Opin Lipidol 1998; 9: 211-9.
Rye KA, Clay MA, Barter PJ. Remodeling of high density lipoproteins by plasma factors. Atherosclerosis 1999; 45: 227-38.
Lambet G, Chase MB, Dugi K, Bensadoun A, Brewer HB Jr., Santamarina-Fojo S. Hepatic lipase promotes the selective uptake of high density lipoprotein-cholesteryl esters via the scavenger receptors B1. J Lipid Res 1999; 40: 1294-1303.
Collet X, Tall AR, Serajuddin H, Guendouzi K, Royer L, Oliveira H, Barbaras R, Jiang XC, Francone OL. Remodeling of HDL by CETP in vivo and by CETP and hepatic lipase in vitro results in enhanced uptake of HDL CE by cells expressing scavenger receptor B-1. J Lipid Res 1999; 40: 1185-93.
Garcia A, Barbaras R, Collet X, Bogyo A, Chap H, Perret B. High-density lipoprotein 3 receptor-dependent endocytosis pathway in a human hepatoma cell line (HepG2). Biochemistry 1996; 35: 13064-71.
Dugi KA, Brandauer K, Schmidt N, Ramacher D, Kreuzer J. Low hepatic lipase activity is a risk factor for coronary artery disease. Atherosclerosis 2000; 151: 126 (Abstract).
Medzour H, Jones R, Dengremont C, Castro G, Maeda N. Hepatic lipase deficiency increases plasma cholesterol but reduces susceptibility to atherosclerosis in apolipoprotein E deficient mice. J Biol Chem 1997; 272: 13570-75.
Busch SJ, Barnhart RL, Martin GA, Fitzgerald MC, Yates MT, Mao SJ, Thomas CE, Jackson RL. Human hepatic triglyceride lipase expression reduces high density lipoprotein and aortic cholesterol in cholesterol-fed transgenic mice. J Biol Chem 1994; 269: 13376-82.
Jansen H, Verhoeven AJM, Sijbrands EJG. Hepatic lipase: A pro- or anti-atherogenic protein? J. Lipid Res. (in press)
Schmolka IR. Poloxamers in the pharmaceutical industry. In: Tarch PJ, ed. Polymers for controlled drug delivery. Boca Raton: CRC Press, 1991:189-215.
Dobiasova M, Frohlich JJ. Assays of lecithin cholesterol acyl transferase. Methods Mol Biol, 1998; 110: 217-30.
Ramaswamy, M., Zhang X, Burt HM, et al. Human plasma distribution of free paclitaxel and paclitaxel associated with diblock copolymers. J Pharm Sci, 1997; 86: p. 460-4.
Havel RJ, E.H., and Bragdon JH, The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest 1955; 34: p. 1345-1353.
Morton R. E and D.B. Zilversmit, Inter-relationship of lipds transferred by the lipid transfer protein isolated from human lipoprotein-deficient plasma. J Biol Chem 1983; 258: 11751-7.
Morton R. E and D.B. Zilversmit, Purification and characterization of lipid transfer protein(s) from human lipoprotein-deficient plasma. J Lipid Res 1982; 23: 1058-67.
Hocquette JF, Graulet B, Olivecrona T. Lipoprotein lipase activity and mRNA levels in bovine tissues. Comp Biochem Physiol B. Biochem Mol Biol 1998; 121: 201-12.
Berard AM, Clerc M, Brewer Jr. B, Santamarina-Fojo S. A normal rate of cellular cholesterol removal can be mediated by plasma from a patient with familial lecithin-cholesterol acyltransferase (LCAT) deficiency. Clin Chimica Acta 2001; 314: 131-39.
Oschry Y, Eisenberg S. Rat plasma lipoprotein: re-evaluation of a lipoprotein system in an animal devoid of cholesteryl ester transfer activity. J Lipid Res 1982; 23: 1099-1106.
Barter PJ, Lally JI. The activity of an esterified cholesterol transferring factor in human and rat serum. Biochem Biophys Acta 1978; 531: 233-36.
Jolley CD, Woollett LA, Turley SD, Dietschy JM. Centripetal cholesterol flux to the liver is dictated by events in the peripheral organs and not by the plasma high density lipoprotein or apolipoprotein A-I concentration. J Lipid Res 1998; 39: 2143-49.
Dietschy JM, Turley SD. Control of cholesterol turnover in the mouse. J Biol Chem 2002; 277: 3804-9.
Osono Y, Woollett LA, Marotti KR et al. Centripetal cholesterol flux from extrahepatic organs to the liver is independent of the concentration of high density lipoprotein-cholesterol in plasma. Proc Natl Acad Sci USA 1996; 93: 4114-9.
Corresponding Author: Kishor M. Wasan, Associate Professor and Chair, Division of Pharmaceutics and Biopharmaceutics, Faculty of Pharmaceutical Sciences, University of British Columbia, 2146 East Mall, Vancouver B.C., Canada V6T 1Z3. kwasan@interchange.ubc.ca
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps
