J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(2):292-301, 2003
Pharmacokinetics of intravenously administered liposomal all-trans-retinoic acid (ATRA) and orally administered ATRA in healthy volunteers.
Bulent Ozpolat, Gabriel Lopez-Berestein1
Department of Bioimmunotherapy, Section of Immunobiology and Drug Carriers, The University of Texas M. D. Anderson Cancer Center, Texas, USAPeter Adamson
Department of Clinical Pharmacology and Therapeutics, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania, USAChauHwei J. Fu
AAI International-Kansas City Bioanalytical Services, Shawnee, Kansas, USAAnthony H. Williams
Aronex Pharmaceuticals Inc., Woburn, Massachusetts, USA.Received 3 June 2003, Revised 8 July 2003, Accepted 5 August 2003
PDF version
Abstract
Purpose: To determine single-and multiple-dose pharmacokinetics of liposomal-all- trans -retinoic acid (Atragen) following intravenous and oral ATRA (Vesanoid) administration in healthy volunteers. METHODS: This was a randomized, prospective, open-label, parallel pharmacokinetic study in which 29 subjects were given 90 mg/m 2 i.v. liposomal (L)-ATRA (16 subjects) every other day or 45 mg/m 2 oral ATRA (13 subjects) daily over 15 days. Pharmacokinetic parameters were assessed on days 1, 9, and 15. RESULTS: Twenty-two subjects (11 in each group) completed the study and were evaluated. Area under the plasma concentration-time curve [AUC(0,¥)] and maximum plasma concentration (Cmax ) of ATRA did not decrease during the 15 days of L-ATRA treatment. However, the oral ATRA regimen resulted in a significant decrease in the AUC(0,¥) and Cmax of 45.3% and 31.8%, respectively, on day 9 as compared with that to day 1 (p< 0.05). In addition, the mean AUC(0,¥) was 13- and 22-fold greater for L-ATRA than for oral ATRA on days 1 and 9, respectively. Despite the significantly higher plasma concentrations after L-ATRA treatment, the side effects of each formulation were similar, except for dermal exfoliation, which was seen in 31% of the subjects after L-ATRA dosing, and abnormal liver function tests that were seen in 23% of the subjects after oral ATRA administration. CONCLUSIONS: These findings suggest that i.v. administration of L-ATRA maintains higher and stable plasma ATRA concentrations than oral ATRA in healthy subjects after repetitive administration. L-ATRA with a favorable pharmacokinetic profile may have potential advantages over oral ATRA and may be more efficacious in the treatment of acute promyelocytic leukemia or other retinoid-responsive cancers.
Introduction
All- trans -retinoic acid (ATRA), a physiologic derivative of vitamin A (retinol), induces complete clinical remissions (CR) in majority of patients with acute promyelocytic leukemia (APL) (1-3). In contrast to the traditional chemotherapeutics, ATRA can selectively induce terminal differentiation and apoptosis of leukemic cells without causing bone marrow hypoplasia or exacerbation of the frequently occurring fatal hemorrhagic syndromes in patients with APL (3,4). ATRA is also effective in vitro against several tumor cell lines and currently being investigated for efficacy in the treatment and prevention of various types of cancer (5,6).
Despite remission rates of up to 90% with oral ATRA, these CRs are short-lived (3 to 5 months) and ATRA monotherapy is not able to maintain long-term remissions in the patients with APL (1,2,7,8) . Thus, ATRA must be combined with cytotoxic chemotherapy to maintain longer CR (2,3,4). One of the major factors limiting the use of oral ATRA as single agent is rapid and progressive decline in plasma ATRA concentrations over time when drug is administered orally on a chronic daily schedule (10-14) . Pharmacokinetic studies have demonstrated that systemic exposure to ATRA as measured by area under curve (AUC) value decreases at least 5-fold and in some patients even returns to baseline levels by day 21 during continuous daily oral administration (10-15). A marked decrease of ATRA levels in plasma is observed within the first week of the treatment in majority of patients (10). Once ATRA levels is decreased, increasing the oral dose cannot elevate plasma ATRA concentration and it remains at lower levels even if the dose is doubled (10,15). The substantially low plasma ATRA concentrations that are inadequate to support differentiation of APL cells have been shown to be associated with recurrence of the disease (10). In addition, about 20% of patients have been shown to have point mutations in the retinoid-binding domain of promyelocytic leukemia-retinoic acid receptor (PML-RARα), a fusion receptor protein expressed in APL cells due to the translocation t(15;17), indicating that genetic mechanisms, in part, may be involved in the inability of ATRA to keep patients in remission (16,17).
Accelerated metabolism of ATRA due to increased expression of cytochrome P450 enzymes, upregulation of the cytoplasmic retinoic acid binding proteins (CRABPs), and decreased absorption of the drug have been implicated for the basis of progressive reduction in the plasma level of the drug during continuous daily oral ATRA treatment (9,10,13,18-20). Recent studies suggested that a marked induction of expression of the highly specific P450 enzyme CYP26 (retinoic acid hydroxylase) after ATRA exposure in liver (up to thousand fold), intestine, skin, and even APL cells plays probably the most important role in autoinduced metabolism of the drug and the rapid elimination of ATRA during the chronic daily therapy (21-26).
Another important drawback of using oral ATRA is its poor bioavailabilty. ATRA is almost insoluble in aqueous solutions and its intestinal absorption is affected by the pH and fatty acid composition of intraluminal bile (27). Therefore, plasma concentrations of ATRA following a p.o. dose are highly variable (28-30).
Liposomes offer a good choice as a drug delivery system and is an alternative method for reducing the toxicity associated with drugs (31-34). Because ATRA is a highly lipophylic molecule, incorporation of ATRA into liposomes provides parenteral formulation. Liposomal incorporation can alter the tissue distribution and pharmacologic profile of drugs (35-40). Most importantly, in preclinical and clinical models when administered intravenously (i.v.), L-ATRA can maintain stable plasma concentrations over a prolonged time after multiple dosing (39,40). Bypassing initial intestinal and hepatic clearance that is associated with repeated oral ATRA administration, L-ATRA may theoretically improve efficacy of the drug (39,41). Furthermore, others and we have previously shown that L-ATRA also provides useful and efficient vehicle for delivering drugs to organs like spleen and bone marrow where the most of the hematopoietic activity and the leukemia cells are located (37,42,43). When incorporated in liposomes, ATRA-associated toxicity is markedly reduced whereas anti-tumor properties (growth inhibition and differentiation induction) of the drug remain the same (44,45). Less toxic form of L-ATRA allows use of higher doses and may potentially lead to higher tissue concentrations and long duration of action at target site. L-ATRA may be also useful alternative in patients with APL who cannot swallow or absorb capsules, patients with nasogastric tube or small children.
In a phase I clinical study, Estey et al. found that chronic administration of L-ATRA in patients with advanced hematological cancers prevented rapid reduction of plasma ATRA concentrations over time and maintained higher ATRA concentrations than oral administration of ATRA did (40). A preclinical study that used animal model of APL and Phase I and II clinical studies demonstrated that L-ATRA as "monotherapy" was highly effective in inducing long term molecular remissions and may be superior anti-APL agent then oral ATRA (46-49).
In this study, we investigated the pharmacokinetics of intravenously administered L-ATRA and orally administered ATRA in healthy normal subjects after repeated chronic dosing. Because the pharmacokinetics of ATRA in patients may be influenced by drugs such as chemotherapeutic agents, steroids, and imidazole antibiotics that can interfere with ATRA metabolism as well as the patient's nutritional status we determined the pharmacokinetics of each formulation in healthy volunteers. Subjects were given either i.v. L-ATRA (90 mg/m2 ) or oral ATRA (45 mg/m2 ) over 15 days to better define the time course of changes following long-term administration of L-ATRA and directly compare the changes with those seen with oral ATRA use. The doses selected for this study were the standard doses used in remission induction regimens in patients with APL.
Subjects and Methods
Twenty-nine healthy human subjects were enrolled in this study from 26 January 2001 to February 10, 2001: 16 were randomly assigned to the i.v. L-ATRA group, while 13 were assigned to the oral ATRA group. All of the subjects were recruited at the New Orleans Center for Clinical Research in New Orleans, LA. This study was approved by the Institutional Review Board, and written informed consent was obtained from each subject. Only subjects with no abnormalities in their medical history, baseline physical examination, vital signs (blood pressure, pulse, respiratory rate, and temperature), hematology counts, blood chemistry, urine analysis, chest x-ray and electrocardiogram were selected. Female subjects had to have a negative pregnancy test, and all of the subjects had to have negative drug and alcohol screening before the study. Use of all vitamin A derivatives was discontinued at least 7 days prior to enrollment in the study.
Study design and treatments
This study was a comparative open-label, randomized, parallel-group study that was designed to determine the comparable pharmacokinetics of oral ATRA (Vesanoid® capsules, Hoffmann La Roche Inc., Nutley, NJ) and i.v. L-ATRA (Atragen®; Aronex Pharmaceuticals, Woburn, MA). Subjects were administered clinically relevant doses of oral ATRA (45 mg/m2 every day) or L-ATRA (90 mg/m2 every other day) for 15 days. These doses were selected because they are standard doses used in remission induction regimens in previous studies. The L-ATRA used was a sterile, lyophilized powder in a 100-cc vial, reconstituted with 0.9% sodium chloride to form a liposomal suspension with a final ATRA concentration of 2 mg/ml. Because ATRA is light sensitive, all procedures were carried out in subdued light and i.v. bags and lines were covered to prevent excess light exposure during infusion. The i.v. infusion was started at approximately 8 A.M. and lasted for 30 minutes. The subjects remained in a nonsupine position during and for 2 h after each infusion of L-ATRA. The oral formulation of ATRA was given in equal doses twice a day. All subjects were given a standard breakfast about 1 h before receiving the morning doses. All other meals were provided at the normal meal times. There were no restrictions on fluid intake. The safety of each form of ATRA was assessed via the collection of clinical laboratory data, measurements of vital signs, and collection of adverse event information for each subject. Adverse effects were graded using the National Cancer Institute Common Toxicity Criteria Manual (version 2.0, 1999).
Subject samples
On days 1, 9, and 15, blood samples were collected from the subjects following oral ATRA dosing and L-ATRA infusion in heparin-coated tubes and protected from light. The tubes were stored on ice and centrifuged within 30 minutes. In the oral ATRA group, samples were obtained before treatment (hour 0) and 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, and 12 h after the treatment. In the L-ATRA group, samples were obtained before treatment (hour 0), and 0.5, 1, 1.5, 2, 4, 6, 9, 12, and 24 h after the treatment. Additional blood samples were collected before and 4 h after treatment on days 5, 7, 11, and 13.
Statistics
To compare the means of all release data and to assess statistical significance between them, either single-factor analysis of variance (ANOVA) or an unpaired two-tailed t-test was carried out at 5% significance level.
Retinoic acid assay for the determination of plasma ATRA concentrations
Blood samples were centrifuged within 30 min of collection at 2500 rpm for 10 min at 4°C, and plasma samples were stored at -70°C. Quantitation of ATRA in human plasma samples was performed using a bioanalytical method with liquid chromatography and tandem liquid chromatography mass spectral detection (LC/MS/MS) which was developed and validated by AAI International (Shawneer, Kansas City, KA) and is a proprietary of the company. This method met the validation criteria defined in the bioanalytical guidance statement issued by the U.S. Food and Drug Administration. In this method, ATRA and the added international standard, arachidonic acid-d8 was extracted from human plasma via a liquid-liquid extraction using methyl t-butyl ether as the extraction solvent. The extract was then subjected to reverse-phase HPLC on a 5-micron, 50 x 4.6-mm Prism column. The analytes were detected using the PE/Sciex API III+ LC/MS/MS system. Quantitation was achieved by monitoring the product ions (m/z 255 for tretinoin and m/z 267 for arachidonic acid-d8) of precursor ions of m/z 299 for ATRA and m/z 311 for arachidonic acid-d8, respectively. The lower limit of quantitation was 10 ng/ml for ATRA and the analytical range was 10 - 1000 ng/ml. The potential interference of metabolite 4-oxo-ATRA, as well as 13- cis -retinoic acid (isotretinoin) and its metabolite 4-oxo-isotretinoin, was evaluated, with no interference found. The overall accuracy rate was 99.6% to 100% for ATRA with the precision rate ranging from 5.2% to 12.7%. In stability evaluation, no significant degradation was observed for ATRA in human plasma at room temperature (up to 5.5 h) or when subjected to freezing and thawing procedures (up to three cycles; -20°C/37VC). No degradation also was observed for extracts stored at 4°C for up to 15.6 h before chromatography. The results of the validation method demonstrated acceptable performance for specificity, selectivity, sensitivity, linearity, accuracy, precision, and stability.
Pharmacokinetic analysis
Pharmacokinetic parameters were assessed using standard noncompartmental analysis. The maximum plasma concentration (Cmax ) of ATRA and the corresponding time to Cmax (tmax ) observed were experimental values. Additionally, AUC (0,¥) was calculated using the equation:
AUC (0, ¥) = AUC (0, t) + [Ct /lz)
in which AUC (0, t) is the area to the last measurable time point ( t ), Ct is the concentration at the last measurable time point, and lz is the apparent first-order elimination rate constant. Accumulation of ATRA was not expected; therefore, AUC (0,¥) was calculated instead of AUC (0, t) during multiple dosing. The apparent elimination half-life (t1/2 ) of ATRA was calculated as ln(2)/lz. Clearance (CL) was calculated as dose/AUC (0,¥) and the apparent steady-state volume of distribution (Vss ) was calculated as [(AUCMC (0, t) /AUC (0,¥))-0.5t τ ]·[dose/AUC (0,¥)], in which AUCMC (0,¥) is the area under the moment curve extrapolated to infinity and t is the duration of the infusion. CL and the Vss were calculated for L-ATRA only.
Statistical analysis
Statistical analyses were conducted using SAS software program (version 6.12, SAS Institute Inc., Cary, NJ) for the Microsoft Windows 95 operating system (Redmont, WA). The equality of various pharmacokinetic parameters on days 1, 9, and 15 was tested using analysis of variance (ANOVA). These analyses were conducted separately for i.v. L-ATRA and oral ATRA, and no corrections for multiple comparisons were included. AUC (0, t), AUC (0,¥), Cmax , CL, and Vss were also analyzed after logarithmic transformation. An ANOVA model that included effects relative to the drug product (treatment), gender, and gender-by-treatment was used to test for differences between the two formulations and between male and female subjects. This model was used to analyze all of the parameters that were measured after both i.v. L-ATRA and oral administration ATRA. AUC (0,¥) and Cmax were expressed per unit dose (mg/m2 ). An unpaired t-test was used to test for sex-related differences in CL and Vss, which were measured post-treatment only in subjects receiving L-ATRA. The differences in the data were considered statistically significant at a p value less than 0.05.
Results
Subject characteristics, compliance and adverse effects
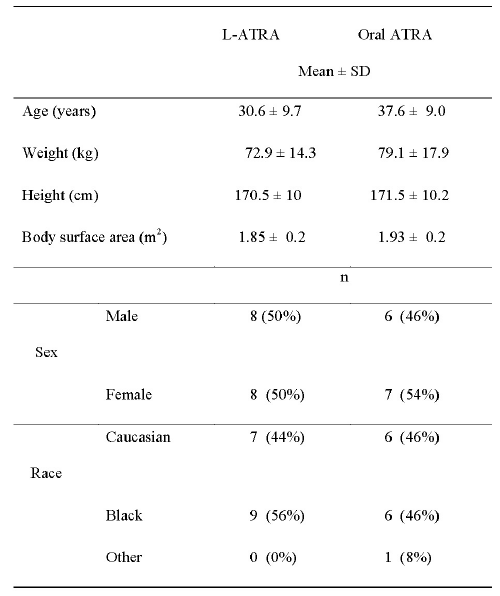
Subject characteristics are summarized in Table 1.
Table 1: Subject characteristics.
The mean age of the subjects in this study was 34.7 ± 9.6 years and ranged 19-46 years. There were no significant differences between the groups in age, weight and height. Eleven evaluable subjects per group completed the study. Two female subjects in the oral ATRA group withdrew from the study on days 3 and 6, respectively, citing moderate (grade 2) side effects of headache, nausea, and vomiting. In addition, three female subjects in the L-ATRA group withdrew from the study on day 4 after receiving their L-ATRA dose because of moderate side effects of nausea, vomiting, and headache. A female subject in the L-ATRA group was withdrawn from the study after the second dose because of escalating hostility toward the staff and other study subjects. In addition, one male subject was withdrawn from the study because he participated in another clinical study.
All of the subjects were evaluated for adverse reactions (Table 2).
Table 2: Adverse reactions.
The majority of the reactions were mild or moderate in both groups. The most common reaction was headache, which occurred in 94% of the subjects in the L-ATRA group and 85% of those in the oral ATRA group. In addition, nausea and vomiting were experienced more often by subjects in L-ATRA group than by those in the oral ATRA group. These effects, including headache, were well tolerated in majority and were easily treated as necessary with acetaminophen, naproxen or promethazine. Five (31%) of the subjects in the L-ATRA group experienced dermal exfoliation, but none of the subjects in the oral ATRA group did. Side effects, such as dryness of skin, constipation and conjunctivitis were seen more often in subjects who received oral ATRA. Interestingly, the results of liver function tests were abnormal in three (23%) subjects in the oral ATRA group, but none of the subjects in the L-ATRA group.
Pharmacokinetics
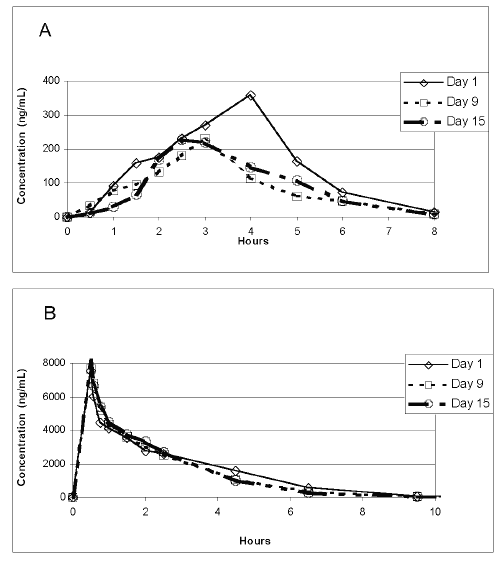
The elimination kinetics of the plasma ATRA concentrations was studied on different days after administration of a single dose of oral ATRA or i.v. L-ATRA to determine whether long-term administration of each formula results in different elimination pattern in healthy subjects. The mean plasma ATRA concentration was 358 ng/ml 4 h after the administration of a single dose of oral ATRA on day 1, ATRA was mostly eliminated from the plasma by 8 h after administration (Figure 1A). However, the mean peak plasma ATRA concentrations decreased significantly to 231.7 ng/ml and 230.6 ng/ml on days 9 and 15, respectively (p< 0.05). The peak plasma concentration after administration of an oral ATRA dose on day 9 was 36.1% lower than that on day 1.
Figure 1: Subjects were administered either oral ATRA (45 mg/m2) or L-ATRA (90 mg/m2) over 15 days. (A) Mean plasma ATRA concentrations following administration of a single dose of oral ATRA on days 1, 9, and 15. (B) Mean plasma ATRA concentrations following administration of a single dose of i.v. L-ATRA administration on days 1, 9, and 15.
The mean plasma concentration of the drug in the subjects who received L-ATRA was calculated on days 1, 9, and 15 (Fig.1 B). The maximum plasma ATRA concentrations rose to 7553.4, 7689, and 8146 ng/ml at the end of the infusion on days 1, 9, and 15, respectively. ATRA was eliminated from the plasma by 12.5 h after L-ATRA infusion; this elimination time did not differ on days 1 and 15, suggesting that L-ATRA display a stable plasma concentration and pharmacokinetic pattern. The elimination of drug from plasma showed a rapid distribution phase followed by a slow elimination phase that lasted until approximately 2 h after the start of the L-ATRA infusion and then a more rapid exponential elimination phase.
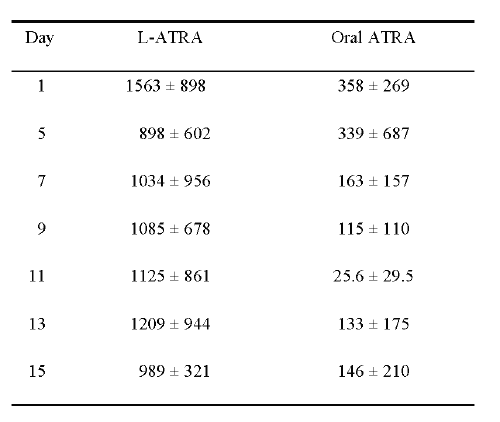
Table 3 shows the mean plasma ATRA concentrations on days 1, 5, 7, 9, 11, 13 and 15, 4-h after administration of oral ATRA or i.v. L-ATRA. The mean plasma ATRA concentrations on day 15 (146 ng/ml) after oral ATRA treatment was about 60% lower than that on day 1 (358 ng/ml), while there was a 37% decrease in the mean ATRA concentration after L-ATRA (1563 ng/ml on day 1 versus 989 ng/ml on day 15).
Table 3: Mean plasma concentrations of ATRA (ng/ml) 4-h after administration (mean ± SD)
The mean plasma ATRA concentrations were found to be 4.4- and 6.7-fold higher after L-ATRA administration than after oral ATRA administration on days 1 and 15, respectively.
Effects of repeated administration of i.v. L-ATRA and oral ATRA on the pharmacokinetics of ATRA
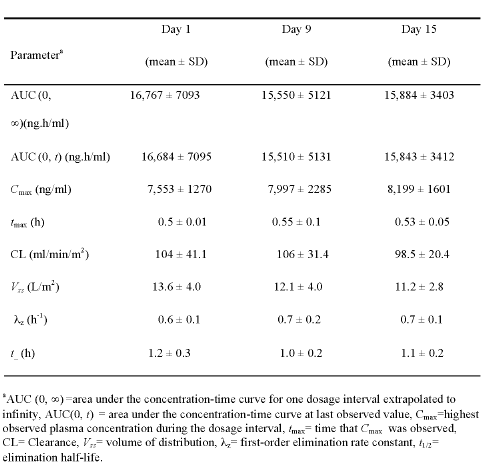
The pharmacokinetic parameters of L-ATRA were evaluated on days 1, 9, and 15; the mean values in the 11 evaluable subjects are summarized in Table 4.
Table 4: Pharmacokinetic parameters in subjects receiving L-ATRA.
The mean AUC (0,¥) of L-ATRA was 16,767 ± 7093 ng.h/ml on day 1, 15,550 ± 5121 ng.h/ml on day 9, and 15,884 ± 3403 ng.h/ml on day 15, indicating that the plasma exposure of ATRA remained constant for up to 2 weeks after repeated administration. The mean Cmax value did not decrease over the 15 days of L-ATRA administration. There was no difference in AUC (0,¥) and Cmax values between male and female subjects.
Because the ATRA concentrations fell below the assay limit (10 ng/ml) before the end of the 48-h dosing interval, there was no accumulation of ATRA with the L-ATRA dosing regimen. Therefore, steady state AUC was not calculated; rather, the AUC (0,¥) was calculated after the doses were given on days 1, 9, and 15. Also, the AUC (0, t) was calculated on days 1, 9, and 15 and was greater than 99% of the AUC (0,¥) in every subject. The mean Cmax value on days 1, 9, and 15 was 7553 ± 1270, 7997 ± 2,285, and 8,199 ± 1,601 ng/ml, respectively, and did not decrease. The other L-ATRA parameters, such as CL, t, λ z , and Vss remained constant after prolonged administration, and did not differ between days 1, 9, and 15 (Table 4). There was also no difference in ATRA CL in male and female subjects who received L-ATRA.
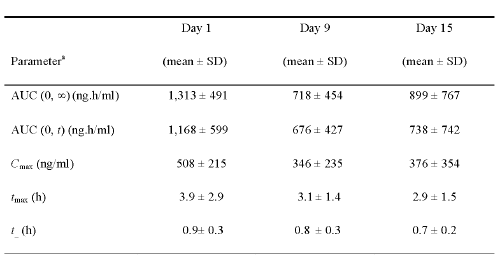
In contrast with L-ATRA, the mean AUC (0,¥) of ATRA significantly decreased within 9 days of the initiation of oral ATRA treatment, falling from 1,313 ± 491 ng.h/ml on day 1 to 718 ± 454 ng.h/ml on day 9 (p< 0.05) (Table 5).
Table 5: Pharmacokinetic parameters in subjects receiving oral ATRA.
The repeated administration of oral ATRA over 9 days resulted in a 45.3% and 31.8 % decrease in the AUC (0, ∞) and Cmax, respectively, when compared with the values on day 1. Similarly, the AUC (0, t) was decreased on day 9 (676 ± 427 ng.h/ml 42% decrease) and day 15 (738 ± 742 ng.h/ml 36.8% decrease) compared with that on day 1 (1,168 ± 599 ng.h/ml). The Cmax also decreased by approximately 30% on days 9 and 15 (346 ± 235 and 376 ± 354 ng/ml, respectively) compared with that on day 1 (508 ± 215 ng/ml). Additionally, the tmax on day 1 (3.9 ± 2.9 h) was significantly longer than on day 9 (3.1 ± 1.4 h) or day 15 (2.9 ± 1.5 h). There was also a significant difference in the overall tmax on days 9 and 15 compared with that on day 1 (p< 0.05).
Discussion
In the present study, we found that i.v. administration of L-ATRA could maintain high plasma concentrations than use of oral ATRA following multiple dosing in healthy volunteers over a period of two weeks. During this period, a significant decrease in plasma ATRA concentrations was observed with oral ATRA but not L-ATRA dosing, suggesting that L-ATRA may prevent rapid decrease in plasma drug concentrations and clearance of drug.
Previous pharmacokinetic studies in patients demonstrated that oral ATRA is unable to sustain steady plasma drug levels during chronic daily administration. In the present study, oral ATRA administration resulted in a significant decrease in the plasma AUC(0,∞) and Cmax by day 9 of treatment (P<0.05). However, AUC(0,∞) and Cmax for L-ATRA did not decrease during the 15 days of treatment. This is probably due to circumvention of accelerated metabolism mediated by P450 enzymes and the hepatic clearance in the liver (39). In fact, we have previously reported that liposomal encapsulation of ATRA decrease metabolism of ATRA by hepatic microsomes in rats that were continuously treated with I.V. L-ATRA (39). However, microsomes isolated from animals that received oral ATRA have shown a significant increase in the metabolism of the ATRA. In the same study, L-ATRA also has produced sustained plasma ATRA concentrations over a prolonged period (7 weeks) when compared to oral ATRA (non-liposomal) which resulted in decreased drug plasma concentrations. In addition, we recently found that L-ATRA induces significantly less P450-CYP26 expression than the same dose of free ATRA in liver cells, suggesting that L-ATRA may prevent induction of a highly specific P450 enzyme and thus preventing autoinduced metabolism of ATRA (50). Taken together, liposomal form of ATRA may avoid adaptations in drug clearance and absorption that may, in part, underlay the observed differences in plasma levels after prolonged administration.
The results of the present study are in agreement with those of a previous phase I clinical trial of L-ATRA in patients with refractory hematological malignancies at M.D. Anderson Cancer Center, which showed no difference in the ATRA AUC values on days 1 and 15 (40). The overall data therefore support the hypothesis that L- ATRA is capable of maintaining consistently high plasma drug concentrations over the course of therapy and may be effective in inducing long-term molecular remissions.
It is important to consider that AUC and Cmax values following L-ATRA dose would be expected to be at least 4 times greater than the oral ATRA dose since a single dose of L-ATRA (90mg/m2) were higher than oral ATRA (22,5 mg/m2 per dose). We found that the mean ATRA AUC value and Cmax was approximately 13 and 15 times higher, respectively, on day 1 and 22 and 23 times higher respectively, on day 9, after L-ATRA administration compared with that after oral ATRA dosing. Because the AUC and Cmax were constant during L-ATRA treatment but decreased during oral ATRA treatment, the ratio of the AUCs for L-ATRA and oral ATRA increased. In addition, The CL of ATRA was consonant with the AUC and Cmax and remained stable during L-ATRA administration. Furthermore, the t1/2 of ATRA measured after L-ATRA infusion was 1.3 times longer on day 1 and 1.6 times longer on day 15 than t1/2 of oral ATRA and even tended to increase during L-ATRA treatment.
After oral administration, ATRA is absorbed in the intestine and carried through the portal vein into the liver where it eventually accumulates. Therefore, due to the first-pass effect significant amount of drug metabolized in the gut and in the liver before it reaches to systemic circulation. We observed considerable interpatient variability in plasma drug exposure following oral administration of ATRA. This may reflect in part interpatient differences in the activity of p450 enzymes such as CYP26 in intestinal and liver cells. In previous studies, decreased gastrointestinal absorption has also been suggested as a source of poor bioavailability of the drug (28,30). In the present study, all subjects received the same standard meal to decrease diet-related variations in absorption. Therefore, all of the statistically significant results (i.e., AUC and Cmax and earlier tmax) after oral ATRA dosing may reflect incomplete bioavailability of the oral formulation and/or induced metabolism of the drug in the gastrointestinal tract and liver (20,22). Based on our findings with L-ATRA, we estimated the bioavailability of oral ATRA to be no more than 25% to 30%. In addition, the plasma CL of ATRA (193 ± 81.0 ml/min) was much lower than the hepatic blood flow; therefore incomplete bioavailability of an oral ATRA cannot be attributed to only first-pass hepatic metabolism. This led us to infer that either intestinal wall metabolism or gut lumen transformation may significantly contribute to the incomplete bioavailability of oral ATRA.
Despite the significantly higher (about 13 to 15 times) plasma ATRA concentrations achieved with L-ATRA administration, its side effects were similar to those of oral ATRA. The majority of the reactions were mild or moderate in both groups. Percentage of patients that had headache, nausea and vomiting were higher in L-ATRA than oral ATRA. Dermal exfoliation was seen only in L-ATRA group but not in oral ATRA group, suggesting L-ATRA tends to accumulate in the skin. In contrast, some of the reactions, such as dryness of skin, constipation and conjunctivitis were seen more in subjects who received oral ATRA. The most importantly, the results of liver function tests were normal in the L-ATRA group but abnormal 23% of subjects in the oral ATRA group at the end of the 15-day treatment schedule, suggesting that L-ATRA is safer in terms of causing hepatotoxicity. In phase I dose-escalation study, the maximum-tolerated dose of L-ATRA was 140 mg/m2 (40). At this dosage, the L-ATRA formulation was well tolerated, with safety profile similar to that of oral ATRA (45 mg/m2). Toxicities observed in the study at dose (90 mg/m2) have been considered as minor and consisted mainly of transient headache (40).
Different treatment modalities have been investigated in attempt to overcome the accelerated metabolism of ATRA that leads to a progressive decline in plasma ATRA concentrations during a chronic daily schedule to improve the outcome of APL. These modalities include the use of inhibitors of cytochrome p450 enzymes, administration of oral ATRA on an intermittent schedule, and encapsulation of ATRA in liposomes (26, 49,51). In these approaches, L-ATRA has been the most successful in terms of delaying or overcoming emerging of accelerated metabolic state that is commonly seen during oral ATRA treatment. One of the rationales for using liposomes as a drug delivery system is to improve the performance of ATRA by its delivery to the specific tissue sites where the disease present. Liposomal formulation allows the use of higher doses without increasing toxicity and potentially lead to higher concentrations in these tissues, such as spleen, liver and bone marrow for prolonged periods; thus potentially yielding longer CR rates in APL patients (36-38).
In conclusion, overall data suggest that plasma levels with L-ATRA, contrary to what is observed with oral ATRA, are maintained even after prolonged administration of L-ATRA. The favorable pharmacokinetic profile of L-ATRA along with the higher consistent plasma exposure to ATRA and safety profile of L-ATRA suggest that L-ATRA has potential advantages over oral ATRA and may be highly effective as a monotherapy for the long-term treatment of APL and other retinoid-sensitive tumors.
References
Fenaux, P., Le Deley, M. C., Castaigne, S., Archimbaud, E., Chomienne, C., Link, H., Guerci, A., Duarte, M., Daniel, M. T., Bowen, D., and . Effect of all transretinoic acid in newly diagnosed acute promyelocytic leukemia. Results of a multicenter randomized trial. European APL 91 Group. Blood, 82: 3241-3249, 1993.
Tallman, M. S., Andersen, J. W., Schiffer, C. A., Appelbaum, F. R., Feusner, J. H., Woods, W. G., Ogden, A., Weinstein, H., Shepherd, L., Willman, C., Bloomfield, C. D., Rowe, J. M., and Wiernik, P. H. All-trans retinoic acid in acute promyelocytic leukemia: long-term outcome and prognostic factor analysis from the North American Intergroup protocol. Blood, 100: 4298-4302, 2002.
Lo, C. F., Nervi, C., Avvisati, G., and Mandelli, F. Acute promyelocytic leukemia: a curable disease. Leukemia, 12: 1866-1880, 1998.
Tallman, M. S. and Nabhan, C. Management of acute promyelocytic leukemia. Curr.Oncol.Rep., 4: 381-389, 2002.
Lotan, R. Effects of vitamin A and its analogs (retinoids) on normal and neoplastic cells. Biochim Biophys Acta. 605:33-91, 1988.
Lotan, R. Retinoids and apoptosis: implications for cancer chemoprevention and therapy. J.Natl.Cancer Inst., 87: 1655-1657, 1995.
Castaigne, S., Chomienne, C., Daniel, M. T., Ballerini, P., Berger, R., Fenaux, P., and Degos, L. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood, 76: 1704-1709, 1990.
Warrell, R. P., Maslak, P., Eardley, A., Heller, G., Miller, W. H., and Frankel, S. R. Treatment of acute promyelocytic leukemia with all-trans retinoic acid: an update of the New York experience. Leukemia, 8: 929-933, 1994.
Warrell, R. P. Retinoid resistance in acute promyelocytic leukemia: new mechanisms, strategies, and implications. Blood, 82: 1949-1953, 1993.
Muindi, J., Frankel, S. R., Miller, W. H., Jakubowski, A., Scheinberg, D. A., Young, C. W., Dmitrovsky, E., and Warrell, R. P. Continuous treatment with all-trans retinoic acid causes a progressive reduction in plasma drug concentrations: implications for relapse and retinoid "resistance" in patients with acute promyelocytic leukemia. Blood, 79: 299-303, 1992.
Lefebvre, P., Thomas, G., Gourmel, B., Agadir, A., Castaigne, S., Dreux, C., Degos, L., and Chomienne, C. Pharmacokinetics of oral all-trans retinoic acid in patients with acute promyelocytic leukemia. Leukemia, 5: 1054-1058, 1991.
Smith, M. A., Adamson, P. C., Balis, F. M., Feusner, J., Aronson, L., Murphy, R. F., Horowitz, M. E., Reaman, G., Hammond, G. D., Fenton, R. M., and . Phase I and pharmacokinetic evaluation of all-trans-retinoic acid in pediatric patients with cancer. J.Clin.Oncol., 10: 1666-1673, 1992.
Muindi, J. R., Frankel, S. R., Huselton, C., DeGrazia, F., Garland, W. A., Young, C. W., and Warrell, R. P. Clinical pharmacology of oral all-trans retinoic acid in patients with acute promyelocytic leukemia. Cancer Res., 52: 2138-2142, 1992.
Trump, D. L., Smith, D. C., Stiff, D., Adedoyin, A., Day, R., Bahnson, R. R., Hofacker, J., and Branch, R. A. A phase II trial of all-trans-retinoic acid in hormone-refractory prostate cancer: A clinical trial with detailed pharmacokinetic analysis. Cancer Chemother Pharmacol., 39: 349-356, 1997.
Adamson, P.C., Balis, F.M, Smith M.A, Murphy R.F, Godwin, K.A, Poplac, D.C. Dose-dependent pharmacokinetics of all-trans-retinoic acid. J Natl Cancer Inst. 84:1332-1340, 1992.
Imaizumi, M., Suzuki, H., Yoshinari, M., Sato, A., Saito, T., Sugawara, A., Tsuchiya, S., Hatae, Y., Fujimoto, T., Kakizuka, A., Konno, T., and Iinuma, K. Mutations in the E-domain of RAR portion of the PML/RAR chimeric gene may confer clinical resistance to all-trans retinoic acid in acute promyelocytic leukemia. Blood, 92: 374-382, 1998.
Shao, W., Benedetti, L., Lamph, W. W., Nervi, C., and Miller, W. H. A retinoid-resistant acute promyelocytic leukemia subclone expresses a dominant negative PML-RAR alpha mutation. Blood, 89: 4282-4289, 1997.
Cormic M, Delva, L, Castaigne, S, Lefebvre, P, balitrand, N, Degos, L, Chomienne, C. in vitro all-trans-retinoic acid sensitivity and cellular retinoic acid binding protein (CRABP) levels in relapse leukemic cells after remission induction. Leukemia; 8:914-921, 1994.
Adamson, P. C., Boylan, J. F., Balis, F. M., Murphy, R. F., Godwin, K. A., Gudas, L. J., and Poplack, D. G. Time course of induction of metabolism of all-trans-retinoic acid and the up-regulation of cellular retinoic acid-binding protein. Cancer Res., 53: 472-476, 1993.
Delva, L., Cornic, M., Balitrand, N., Guidez, F., Miclea, J. M., Delmer, A., Teillet, F., Fenaux, P., Castaigne, S., Degos, L., and . Resistance to all-trans retinoic acid (ATRA) therapy in relapsing acute promyelocytic leukemia: study of in vitro ATRA sensitivity and cellular retinoic acid binding protein levels in leukemic cells. Blood, 82: 2175-2181, 1993.
Yamamoto Y, Zolfaghari R, Ross A.C .Regulation of CYP26 (cytochrome P450RAI) mRNA expression and retinoic acid metabolism by retinoids and dietary vitamin A in liver of mice and rats. FASEB J 14(13):2119-27, 2000.
Ozpolat, B., Tari, A. M., Mehta, K., and Lopez-Berestein All-trans-Retinoic Acid (ATRA) induces expression of cytochome P450RAI (CYP26) in human Intestinal, Liver and Promyelocytic Leukemia cells: A potential mechanism contributing to increased ATRA metabolism and resistance. Proceedings of AACR (Blood), 92: 374, 2000.
Marikar, Y., Wang, Z., Duell, E. A., Petkovich, M., Voorhees, J. J., and Fisher, G. J. Retinoic acid receptors regulate expression of retinoic acid 4- hydroxylase that specifically inactivates all-trans retinoic acid in human keratinocyte HaCaT cells. J.Invest Dermatol., 111 : 434-439, 1998.
Lampen, A., Meyer, S., Arnhold, T., and Nau, H. Metabolism of vitamin A and its active metabolite all-trans-retinoic acid in small intestinal enterocytes. J.Pharmacol.Exp.Ther., 295: 979-985, 2000.
Ozpolat, B., Mehta, K., Tari, A. M., and Lopez-Berestein, G. All-trans-Retinoic acid-induced expression and regulation of retinoic acid 4-hydroxylase (CYP26) in human promyelocytic leukemia. Am.J.Hematol., 70: 39-47, 2002.
Miller, W. H. The emerging role of retinoids and retinoic acid metabolism blocking agents in the treatment of cancer. Cancer, 83: 1471-1482, 1998.
Regazzi, M. B., Iacona, I., Gervasutti, C., Lazzarino, M., and Toma, S. Clinical pharmacokinetics of tretinoin. Clin.Pharmacokinet., 32: 382-402, 1997.
Adamson, P. C., Pitot, H. C., Balis, F. M., Rubin, J., Murphy, R. F., and Poplack, D. G. Variability in the oral bioavailability of all-trans-retinoic acid. J.Natl.Cancer Inst., 85: 993-996, 1993.
Lazzarino, M., Regazzi, M. B., and Corso, A. Clinical relevance of all-trans retinoic acid pharmacokinetics and its modulation in acute promyelocytic leukemia. Leuk.Lymphoma, 23: 539-543, 1996.
Rigas, J. R., Francis, P. A., Muindi, J. R., Kris, M. G., Huselton, C., DeGrazia, F., Orazem, J. P., Young, C. W., and Warrell, R. P. Constitutive variability in the pharmacokinetics of the natural retinoid, all-trans-retinoic acid, and its modulation by ketoconazole. J.Natl.Cancer Inst., 85: 1921-1926, 1993.
Gregoriadis, G. and Florence, A. T. Liposomes and cancer therapy. Cancer Cells, 3: 144-146, 1991.
Lasic, D. D. and Papahadjopoulos, D. Liposomes revisited. Science, 267: 1275-1276, 1995.
Torchilin, V. P. Drug targeting. Eur.J.Pharm.Sci., 11 Suppl 2: S81-S91, 2000.
Harrington, K. J., Syrigos, K. N., and Vile, R. G. Liposomally targeted cytotoxic drugs for the treatment of cancer. J.Pharm.Pharmacol., 54: 1573-1600, 2002.
Lopez-Berestein, G., Rosenblum, M. G., and Mehta, R. Altered tissue distribution of amphotericin B by liposomal encapsulation: comparison of normal mice to mice infected with Candida albicans. Cancer Drug Deliv., 1: 199-205, 1984.
Lopez-Berestein, G., Bodey, G. P., Frankel, L. S., and Mehta, K. Treatment of hepatosplenic candidiasis with liposomal-amphotericin B. J.Clin.Oncol., 5: 310-317, 1987.
Lopez-Berestein, G., Kasi, L., Rosenblum, M. G., Haynie, T., Jahns, M., Glenn, H., Mehta, R., Mavligit, G. M., and Hersh, E. M. Clinical pharmacology of 99mTc-labeled liposomes in patients with cancer. Cancer Res., 44: 375-378, 1984.
Lopez-Berestein, G., Rosenblum, M., Sadeghi, T., and Mehta, K. Pharmacokinetics, tissue distribution, and toxicity of tretinoin incorporated in liposomes. J Liposome Res, 4: 689-700, 1994.
Mehta, K., Sadeghi, T., McQueen, T., and Lopez-Berestein, G. Liposome encapsulation circumvents the hepatic clearance mechanisms of all-trans-retinoic acid. Leuk. Res., 18: 587-596, 1994.
Estey, E., Thall, P. F., Mehta, K., Rosenblum, M., Brewer, T., Simmons, V., Cabanillas, F., Kurzrock, R., and Lopez-Berestein, G. Alterations in tretinoin pharmacokinetics following administration of liposomal all-trans retinoic acid. Blood, 87: 3650-3654, 1996.
Mehta, K., Estey, E., and Lopez-Bernstein, G. Liposome-Mediated Delivery of Retinoids. In G. Gregoriadis and McCormack (eds.), Targetting of Drugs: Strategies for Stealth Therapeutic Systems, pp. 87-94. New York: Plenum press, 1998.
Kasi, L. P., Lopez-Berestein, G., Mehta, K., Rosenblum, M., Glenn, H. J., Haynie, T. P., Mavligit, G., and Hersh, E. M. Distribution and pharmacology of intravenous 99mTc-labeled multilamellar liposomes in rats and mice. Int.J.Nucl.Med.Biol., 11: 35-37, 1984.
Gray, A. and Morgan, J. Liposomes in haematology. Blood Rev., 5: 258-272, 1991.
Drach, J., Lopez-Berestein, G., McQueen, T., Andreeff, M., and Mehta, K. Induction of differentiation in myeloid leukemia cell lines and acute promyelocytic leukemia cells by liposomal all-trans-retinoic acid. Cancer Res., 53: 2100-2104, 1993.
McCarthy, D., Dollar, G., and Hill, D. Toxicity and antitumor activity of liposome entrapped retinoid Ro13-7410. Selective Cancer Therapy, 7: 151, 1992.
Westervelt, P., Pollock, J. L., Oldfather, K. M., Walter, M. J., Ma, M. K., Williams, A., DiPersio, J. F., and Ley, T. J. Adaptive immunity cooperates with liposomal all-trans-retinoic acid (ATRA) to facilitate long-term molecular remissions in mice with acute promyelocytic leukemia. Proc.Natl.Acad.Sci.U.S.A, 99: 9468-9473, 2002.
Estey, E. H., Giles, F. J., Kantarjian, H., O'Brien, S., Cortes, J., Freireich, E. J., Lopez-Berestein, G., and Keating, M. Molecular remissions induced by liposomal-encapsulated all-trans retinoic acid in newly diagnosed acute promyelocytic leukemia. Blood, 94: 2230-2235, 1999.
Douer, D., Estey, E., Santillana, S., Bennett, J. M., Lopez-Bernstein, G., Boehm, K., and Williams, T. Treatment of newly diagnosed and relapsed acute promyelocytic leukemia with intravenous liposomal all-trans retinoic acid. Blood, 97: 73-80, 2001.
Ozpolat B, Lopez-Berestein G.Liposomal-all-trans-retinoic acid in treatment of acute promyelocytic leukemia. Leuk Lymphoma,43(5):933-41, 2002
Ozpolat, B., Tari, A. M., Mehta, K., and Lopez-Berestein, G. Expression and Regulation of a cytochRome P450 Enyzme (CYP26) by All-trans-retinoic acid (ATRA) and Vitamin A in human Liver, Intestinal and Endothelial cells (Abstract). Proceedings of AACR (Blood), 2003.
Adamson, P. C., Bailey, J., Pluda, J., Poplack, D. G., Bauza, S., Murphy, R. F., Yarchoan, R., and Balis, F. M. Pharmacokinetics of all-trans-retinoic acid administered on an intermittent schedule. J.Clin.Oncol., 13: 1238-1241, 1995.
Corresponding Author: Gabriel Lopez-Berestein, Section of Immunobiology and Drug Carriers, Department of Bioimmunotherapy, Box 422, The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX, USA 77030. glopez@ mdanderson.org
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps