J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(2):274-281, 2003
Preparation and in vitro evaluation of chitosan matrices for colonic controlled drug delivery.
Ylenia Zambito, Giacomo Di Colo1
Department of Bioorganic Chemistry and Biopharmaceutics, University of Pisa, Pisa, ItalyReceived 3 June 2003, Revised 19 June 2003, Accepted 05 August 2003
PDF version
Abstract
PURPOSE. The work was aimed at studying in vitro the release of 5-aminosalicylic acid (5-ASA) or diclofenac sodium (DS) from matrices based on chitosan (Ch) or Ch hydrochloride (Ch-HCl), destined to be introduced into enteric-coated capsules for controlled release to the colon. METHODS. Matrices (diameter, 6 mm; weight, 50 mg) were prepared by compression of Ch or Ch-HCl microparticles mixed with 20 % 5-ASA or DS powder. Drug release from matrices to isotonic neutral buffers of different molarity was studied in vitro. In some cases, matrix incubation in rat cecal contents preceded the release test. RESULTS. The matrices, especially the Ch-HCl-based ones, swelled in the dissolution medium without disintegrating. Drug release was diffusion-controlled and followed square-root-time kinetics. Release depended on the pH-dependent aqueous solubility of the drug. The internal pH of the swollen Ch-HCl-based matrix was acidic, so 5-ASA solubility and release were influenced by penetration of salts from the external buffer. In the Ch-HCl-based matrix DS was converted into the scarcely soluble diclofenac free acid, which prolonged the time for release of 50 % dose excessively (t50 =11.26 h). The enzymatic action of rat cecal microflora accelerated drug release from the Ch-HCl-based matrix. On the other hand, neither such a microflora nor the external medium hydrodynamics significantly affected drug release from the Ch-based matrix. CONCLUSIONS. The Ch-based matrix was a reliable colonic controlled-release system for 5-ASA (t50 =1.97 h) or DS (t50 =3.58 h). For in vivo application, a number of matrices adequate to make up the therapeutic drug dose should be introduced into enteric-coated size 00 capsules.
INTRODUCTION
A great deal of research work has been devoted to specific drug delivery to the colon, as documented by the numerous publications recently reviewed by Yang et al. (1). Indeed, colon-specific delivery with a controlled release pattern provides more effective therapy for such chronic diseases as irritable bowel syndrome and inflammatory bowel disease, including Crohn's disease and ulcerative colitis. A colon-specific drug delivery system releasing the drug at controlled rates should prevent drug release in the stomach and small intestine, and start releasing the drug upon entry into the colon. Cole et al. (2) showed that hydroxypropylmethylcellulose (HPMC) capsules, enteric-coated with Eudragit® FS 30 D by an industrially viable process, disintegrated towards the distal small intestine and proximal colon. The authors intended their capsules as containers for colon-specific drug administration. Nevertheless, capsules of this type could profitably be used to carry a controlled-release device across the upper GI tract and to prevent drug release in this tract. The device would be released intact from the capsule in the proximal colon, where it would start releasing the drug. By these means, controlled-release systems of whatever type could be targeted to the colon, provided they were fit to enter the capsule. Along this line, this paper presents a study of controlled-release systems for diclofenac sodium (DS) and 5-aminosalicylic acid (5-ASA), intended to be conveyed to the colon by the above vehicle. 5-ASA is known to be the active therapeutic moiety of sulfasalazine (3), a prodrug used for the treatment of ulcerative colitis and Crohn's disease. Since as many as 30 % patients are unable to tolerate treatment with sulfasalazine, due to side effects and toxicity caused by its metabolite sulfapyridine (4), it is desirable to deliver 5-ASA specifically to the colon with low toxicity carriers. DS is widely used in the treatment of chronic inflammatory diseases (5). Due to the rapid systemic clearance of this drug (6, 7) repeated daily dosing is required. This warrants the use of sustained-release formulations to improve patient compliance (8). Since DS is well absorbed in the colon (9), colon-targeted delivery systems are particularly advisable, as they would avoid drug release in the stomach, thus eliminating local side effects (10, 11). Chitosan (Ch) was chosen as the release-controlling polymer in virtue of its well-known biocompatibility and biodegradability. Ch has extensively been used to sustain and control drug delivery (see, e.g., the review of Singla and Chawla, (12)). However, to the best of our knowledge still lacks literature information on drug release from compressed matrix tablets of plain, chemically unmodified Ch. In fact, the tablet formulations described in the literature contained additional ingredients to facilitate compaction, since commercial Ch is fibrous in nature, and therefore, difficult to compact. Sabnis et al. (13) depolymerised commercially available Ch and exploited the increased microcrystallinity and consequent compactability of the depolymerisation product to prepare a sustained-release tablet formulation of DS by direct compression of powders. In the present work, commercial Ch was converted into microspheres of Ch hydrochloride (Ch-HCl) or Ch freebase, which could be directly compressed, together with the drug, into matrix tablets of 6-mm diameter. These were fit to be introduced into size 00 HPMC capsules, enteric-coated by the process described by Cole et al. (2). The release of DS or 5-ASA from such matrices and the susceptibility of drug release to the polymer degrading action of the cecal microflora were evaluated in vitro using rat cecal microflora, in virtue of the similarity with human intestinal microflora (14). During matrix transit through distal small intestine and colon the pH of the matrix environment, although variable, should keep above the pKa of Ch, that is, 5.6. Then the polymer should stay in, or be converted into the insoluble free base form, irrespective of the physiologic pH variations. Therefore, in the present in vitro experiments, aimed at evaluating the release-controlling properties of matrices, neutral buffers were used as dissolution media.
mATERIALS AND mETHODS
Materials
DS, 5-ASA, Ch (from Crab Shells, minimum 85% deacetylated) were purchased from Sigma, Milano, Italy. Sodium chloride, sodium dihydrogen phosphate, potassium dihydrogen phosphate and di-sodium hydrogen phosphate dihydrate were purchased from Carlo Erba, Milano, Italy. Potassium chloride, calcium chloride dihydrate, magnesium chloride hexahydrate and potassium hydroxide pellets were purchased from Fluka, Buchs, Switzerland. Methanol GR was purchased from Merck, Darmstadt, Germany. All other chemicals and solvents were of reagent grade. The materials were used as received.
Characterisation of commercial Ch
Commercial Ch was characterised by determining its molecular weight by capillary viscometry (Ostwald capillary viscometer, Series 200) and its deacetylation degree by IR spectroscopy (Mattson 3000 FTIR spectrophotometer, Mattson Instruments, Madison, WI, USA), following the procedures described by Khalid et al. (15). The water weight fraction contained in the commercial Ch was determined by desiccation at 100°C.
Preparation of Ch-HCl microspheres
Ch-HCl microspheres of < 2.5 μm diameter were obtained by spray drying a 0.5 % Ch-HCl aqueous solution prepared from a suspension of 6 g commercial Ch in 800 ml water, as described previously (16). Ch was almost completely dissolved by bringing the suspension to pH 4.7 by 21.8 ml of 1N HCl. After filtration to remove traces of non-dissolved material, a small aliquot of solution was evaporated to dryness and the residue was weighed, after equilibration with the room atmosphere, to determine the actual Ch-HCl concentration. Then the solution was diluted to a concentration of 0.5 % and spray-dried. Following preparation, the microspheres were allowed to equilibrate with the room atmosphere before use (water content, around 11 %).
Preparation of Ch microspheres
Microspheres of Ch freebase were obtained by treatment of Ch-HCl microspheres with methanolic KOH. Ch-HCl microspheres (1 g) were suspended in methanol (200 ml), then, a volume of 0.5 M methanolic KOH calculated to neutralise the HCl contained in the microspheres (6.4 ml) was gradually added under stirring. The number of KOH equivalents was calculated based on the HCl equivalents added to Ch to obtain the Ch-HCl microspheres, as described above. The resulting Ch microspheres were collected by filtration under vacuum (0.45 μm pore size membrane filter), rinsed with methanol and brought to a constant weight by an air stream at room temperature. The size and shape of microspheres, as observed by an optical microscope, were unchanged compared to the parent Ch-HCl microspheres.
Preparation of matrices
Matrices of 50 mg weight and 6 mm diameter, containing 20 % w/w DS or 5-ASA, were obtained by compressing blends of DS or 5-ASA (particle size < 106 mm) with Ch-HCl or Ch microspheres. Mixing of ingredients was carried out manually, by means of a spatula. The comparatively high drug weight fraction in matrices was meant to minimise the number of matrices needed to make up the therapeutic dose. The compaction force (9800 N) was applied by a Perkin-Elmer hydraulic press.
Release experiments
An already described method was used to measure the drug release kinetics from the matrices containing DS or 5-ASA (17, 18). The method was a modification of the USP rotating basket method, designed to realise strictly controlled hydrodynamics of the matrix environment. The matrix was placed into a stainless steel wire mesh bag, fastened to a stainless steel hook, fixed to the shaft of a glass paddle stirrer, in such a way that the matrix was held under the impeller, at a 1.2-cm distance from the paddles. At time t=0, the stirrer, with attached matrix, was immersed in 100 ml of phosphate buffer (PB) pH 7.4, 0.13 M (isotonic) or, in some cases, of PB pH 7.4, 0.0325 M, made isotonic with NaCl. The buffer was contained in a jacketed beaker (internal diameter, 6.5 cm; internal height, 9.0 cm) and maintained at 37±0.1°C by a thermostat. The matrix was placed at the centre of the dissolution medium, and stirring at 150 rpm or, in some cases, 60 rpm was initiated by means of a synchronous motor. At pre-established time intervals, 10-ml dissolution medium samples were withdrawn and replaced by an equal volume of fresh, pre-thermostated medium. The withdrawn samples were analysed spectrophotometrically (Hitachi 150-20 spectrophotometer, Tokyo, Japan) at 276 nm, for DS, or 330 nm, for 5-ASA. Blank runs showed the absence of significant interference with the spectrophotometric measurements. Since the 5-ASA or DS concentration in the dissolution medium was always much less than one tenth of the drug solubility in such a medium, sink conditions could be assumed.
Evaluation of matrix material interaction with the dissolution medium salts
Drug-free tablets made of Ch-HCl were maintained 24 h in contact with pH 7.4, 0.13 M PB under the conditions of the release experiments described above. After washing with water and drying to constant weights by an air stream at room temperature, the tablets were powdered in a ball mill, and then an accurately weighed (10-5 g) powder amount (50-70 mg) was stirred overnight in 100 ml water in order to determine the fraction of water-soluble material. Then, the insoluble residue was collected by centrifugation, dried to a constant weight by an air stream at room temperature and weighed.
Evaluation of susceptibility of drug release to cecal microflora
Such susceptibility was evaluated by determining the effect of matrix incubation in rat cecal contents on the drug release kinetics. A suspension of 33 % w/v cecal contents in bicarbonate buffer pH 7 (BCB) was prepared as described by Tozaki et al. (19). Each of three matrices based on Ch-HCl or Ch, containing 20 % w/w 5-ASA, was introduced into a screw-capped glass tube containing 6 ml of the above suspension. The tube was immediately fixed in radial position to a wheel mixer rotating vertically at 26 rpm in a thermostatic oven at 37°C. After 4 h of incubation, each matrix was rapidly and thoroughly flushed with BCB, then the kinetics of drug release from matrix were determined over 2.5 h, using pH 7.4, 0.13 M PB as the dissolution medium. For reference, for each matrix a blank run was carried out, where an equal matrix was incubated in BCB free of cecal contents, after which the drug release kinetics were determined as usual. Following the release measurements, the matrices were fully depleted of drug by keeping them in contact with the dissolution medium for 24 h. In this way, the drug amount contained in matrix at time zero of the kinetic measurements were assessed, and hence, the percentage released in time t could be calculated.
Solubility determinations
Excess drug was shaken in the appropriate buffer solution at 37°C. Periodically, aliquots of suspension were withdrawn, filtered (0.45 mm pore size) and analysed spectrophotometrically, until equilibrium was attained. In all cases, this required less than 8 h. Each drug was analysed at pH 7.4 at the respective wavelengths indicated in the Release experiments Section. 5-ASA was also analysed at pH 4.5, at a wavelength of 297 nm. The solubility characteristics of DS at pH 4.5 were determined as follows. A 0.7 % DS aqueous solution was acidified to pH 4.5 with HCl, which resulted in an immediate precipitation of diclofenac free acid (DH). The suspension was shaken at 37°C and periodically analysed spectrophotometrically at 276 nm, after filtering (0.45 mm pore size). The equilibrium concentration was attained in less than 8 h.
rESULTS AND dISCUSSION
Characterisation of commercial Ch
The average viscometric molecular weight of commercial Ch and its deacetylation degree, determined by the methods described by Khalid et al. (15), were 1460 kDa and 89.9 %, respectively. The commercial Ch contained 12.3 % water.
Behaviour of matrices in the dissolution medium
Either the Ch-HCl or Ch microspheres, mixed with either 5-ASA or DS powder could easily be compacted into mechanically stable matrices, without the aid of any additional ingredient. Once in contact with the dissolution medium the matrices showed no visible disintegration but rather, they showed an apparent volume increase, due to water absorption, which was sensibly more marked for the matrices based on Ch-HCl. The test intended to evaluate the interaction of the Ch-HCl matrices with the dissolution medium salts showed that, after 24 h residence of the Ch-HCl tablets in the dissolution medium of the release experiments, only 57.7 % of the tablet material was water-soluble. Since Ch-HCl is water-soluble, these results indicated that the buffer salts could penetrate into the water-swollen tablets and partially converted the water-soluble, acidic Ch-HCl into the insoluble Ch free base. Such a penetration of buffer salts could influence the solubility of 5-ASA or DS within the swollen matrix, since, as appears from the data in Table 1, the aqueous solubility of either drug is pH-dependent.
Table 1: Solubility values.
Release studies
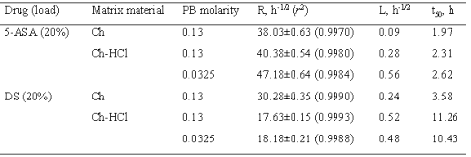
The release data for the matrices under study are presented in Figures 1 and 2 as plots of percent dose released vs. square root of time. In all cases, a major portion of plot was linear, hence the release kinetics could be described by a rate parameter, R, that is the slope of the straight line, and a lag time parameter, L, that is the ü t-axis intercept. For each plot, linear regression analysis was extended to the set of data points that gave the best fit, as judged from the r2 value. The parameters for the different matrix types and dissolution medium molarity values are found in Table 2. Also listed in the table are the respective values of t50 that is the time for release of 50 % of the dose.
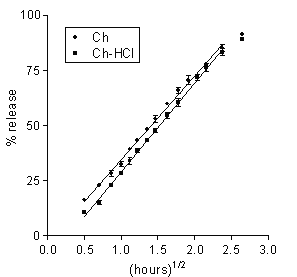
Figure 1: Data on 5-ASA release to 0.13 M PB from Ch-based or Ch-HCl-based matrices loaded with 20 % drug. Each data point is the mean ± SD of at least three values.
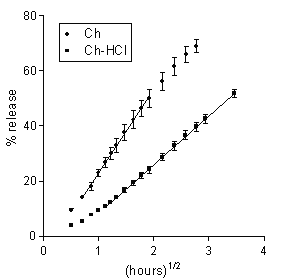
Figure 2: Data on DS release to 0.13 M PB from Ch-based or Ch-HCl-based matrices loaded with 20 % drug. Each data point is the mean ± SD of at least three values.
Table 2: Release parameters: R, rate; L, lag time; t50, time for release of 50 % dose.
Release of 5-ASA
Data on 5-ASA release from the Ch- or the Ch-HCl-based matrices are presented in Figure 1 and Table 2. A linear pattern of both plots up to about 80 % dose released appears from the figure and from the relevant r 2 values in the table. Such square-root-time kinetics suggest that release from both matrix types was governed by drug dissolution and diffusion in the aqueous path created in matrix by polymer hydration (20). The lag time parameters found in Table 2 are to some degree related to the time needed for full polymer hydration. The Ch-HCl matrix apparently swelled to a much higher degree compared to the Ch-based matrix, then the comparatively high porosity of the resulting gel should have allowed faster drug diffusion. However, the rates of 5-ASA release from both matrix types to pH 7.4, 0.13 M PB were similar. This apparent contradiction can be solved if it is admitted that, in addition to diffusivity, solubility in the swollen matrix was a major rate-limiting factor. If the drug had been in the suspended state, indeed, its solubility could determine the concentration gradients in matrix, and hence, the release rate (20). In fact, 5-ASA has limited water solubility, and its concentration in matrix was so high as to suggest that the drug should be suspended in the matrix region where diffusion occurred. As appears from the relevant values in Table 1, 5-ASA solubility is higher at higher pH, therefore it should be higher in the Ch-based matrix, the internal pH of which is thought to be higher than that of the Ch-HCl-based one. Thus, the similar release rates for the Ch- and the Ch-HCl-based matrices can be explained by admitting that the lower drug diffusivity in the less porous Ch-based matrix was counterbalanced by the higher drug solubility. Since Ch-HCl is water-soluble, a possible contribution of matrix erosion to drug release from the Ch-HCl-based matrix had to be ascertained. To this purpose, at the end of the release experiment the matrix was dried and weighed. Taking into account the weight of released drug, no significant polymer loss resulted from a comparison between initial and final weights. Probably, surface erosion of matrix was hindered by the conversion of Ch-HCl into Ch at the matrix-PB interface. As stated above, the 5-ASA solubility in the Ch-HCl-based matrix should be lower than in the Ch-based one. Penetration of buffer salts into the former could cause some gradual drug solubility increase at the early stages of the release process. This was probably responsible for the higher lag time value for the Ch-HCl-based with respect to the Ch-based 5-ASA matrix that appears in Table 2. In fact, as can be observed in the table, the lag time for the former matrix was further increased when the buffer molarity in the dissolution medium was substantially decreased, which can be ascribed to a slower penetration of buffer salts, causing a slower increase of drug solubility within matrix.
Release of DS
The similarity of the aqueous solubility values at pH 7.4 for DS and 5-ASA, seen in Table 1, anticipates a similar release pattern for the two drugs from the Ch-based matrix. Indeed, as appears from a comparison of the respective data in Figures 1, 2 and Table 2, release in both cases followed square-root-time kinetics. The rate parameter for DS is lower, because of its lower solubility, and the lag time parameter for 5-ASA is lower because the higher osmotic power of this drug caused a faster matrix hydration. The data in Table 2 for DS show that the rate parameter for the Ch-HCl-based matrices is considerably lower than that for the Ch-based ones and that the lag time value for the former is considerably higher than that for the latter. Drug solubility in matrix is thought to be a major factor determining such differences. Indeed, matrix hydration elicited an acid-base reaction within matrix between Ch-HCl and DS, which partially converted the former into Ch and the latter into the very little soluble free acid (DH). The very low solubility of DH, reported in Table 1, is believed to slow down both matrix hydration and drug release. With DS as the drug and Ch-HCl as the matrix material, release was not affected by any penetration of buffer salts from dissolution medium into matrix. This can be deduced from the absence of any significant effect of an ample variation of buffer molarity on the relevant kinetic parameters, seen in Table 2.
Effects of matrix environment on release
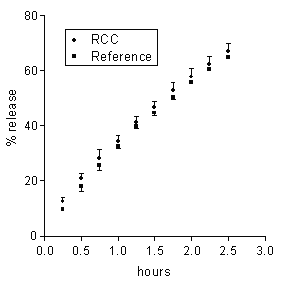
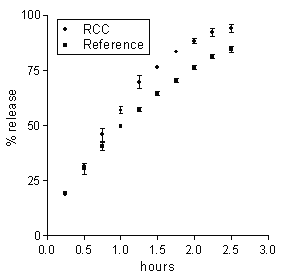
The neutral pH of the receiving buffer was the only property of the colonic environment that was mimicked in the present release experiments. Then, in order for the in vitro release data to be predictive of the in vivo behaviour, such properties of the matrix environment as hydrodynamics and chemical composition should not influence the release properties of matrices. In order to ascertain the relevance of hydrodynamics, the agitation intensity of the external medium was varied. In no case of the 5-ASA and DS matrices based on either Ch or Ch-HCl was release significantly affected by a marked reduction of the agitation intensity from 150 to 60 rpm (data not reported). From here it is inferred that the dissolution medium behaved as a perfect sink, irrespective of its hydrodynamics, because the unstirred aqueous diffusion layer adjacent to the matrix surface was much more permeable to either drug than the matrix itself (20). Drug release from the present matrices to the colon could be influenced by the colonic contents, some components of which could penetrate into matrix and affect some rate-determining factors. Colonic salts could penetrate into the Ch-HCl-based 5-ASA matrix and affect drug release by increasing the internal matrix pH. Besides, colonic bacterial enzymes could penetrate into matrix and degrade the matrix material (19, 21). In order to investigate the susceptibility of drug release to the enzymatic action of the intestinal microflora, each of 5-ASA matrices, based on either Ch or Ch-HCl, was incubated in 6 ml of a medium containing 33 % rat cecal contents. Subsequently, the effect of incubation on drug release was assessed by the usual technique. This procedure, using the cecal contents of one rat for testing each matrix, considerably reduced the need of animals with respect to using a 33 % suspension of cecal contents as the medium of the release tests, as reported by Tozaki et al. (19). Rat cecal microflora was used because of the similarity with human intestinal microflora (14). The drug amount released over the incubation term was 3.6±0.2 or 3.1±0.2 mg for the Ch-HCl or Ch matrices incubated in plain BCB, and 3.4±0.2 or 3.3±0.2 mg for the corresponding matrices incubated in the suspension of cecal contents. From these values it is inferred that essentially the same drug amount was released from each matrix over the incubation term, irrespective of the presence or absence of cecal contents in the incubation medium. It is seen in Figure 3 that 5-ASA release data obtained from Ch-based matrices after incubation in the medium containing rat cecal contents were not significantly different from those obtained from the reference matrices incubated in the medium free of cecal contents.
Figure 3: Data on 5-ASA release from a Ch-based matrix loaded with 20 % drug following 4 h incubation in BCB containing rat cecal contents (RCC) or in plain BCB (reference). Each data point is the mean ± SD of three values.
Tozaki et al. (19) documented a degradation of Ch capsules by rat cecal contents, which promoted insulin release from capsules. The present data indicate that the degrading action upon the Ch-based matrices did not significantly modify any release-determining factors. On the other hand, in the case of the Ch-HCl-based matrix the difference in release data between the matrices incubated in cecal contents and the reference matrices, although small, was beyond the experimental variability, as appears in Figure 4.
Figure 4: Data on 5-ASA release from a Ch-HCl-based matrix loaded with 20 % drug following 4 h incubation in BCB containing rat cecal contents (RCC) or in plain BCB (reference). Each data point is the mean ± SD of three values.
In this case, the enzymatic action presumably caused some increase of matrix porosity, probably because the enzymes could penetrate into the water-swollen matrix.
cONCLUSIONS
Matrices for colonic controlled release of 5-ASA or DS were prepared by direct compression of Ch-HCl or Ch microparticles mixed with drug powders, without the aid of any additional ingredients. Drug release from both matrix types to the neutral buffers used as dissolution media was diffusion-controlled and followed square-root-time kinetics. The matrices based on Ch-HCl swelled without eroding. The acidic pH of the resulting gel converted DS into the scarcely soluble DH, which was released at excessively low rates. It was shown that the high gel porosity could allow penetration of salts and enzymes from the colonic contents into matrix. The salts would tend to neutralise the gel pH, while the enzymes would degrade the polymer, thus increasing matrix porosity. Both processes could affect drug release. Such an influence of the matrix surroundings on drug release implies possible differences between in vitro and in vivo release from the Ch-HCl-based matrices. On the other hand, the release data obtained with the Ch-based matrices are thought to be more predictive of the in vivo performance of such systems. Indeed, they were not significantly affected by those properties of the colonic environment that were not matched by the external phase of the in vitro release experiments, such as hydrodynamics or enzymatic activity. In addition, the external buffer molarity should not be of any relevance, since Ch free base is not expected to react with the buffer salts. For in vivo application, a number of matrices adequate to make up the therapeutic drug dose should be introduced into enteric-coated size 00 HPMC capsules and, thereby, conveyed to the specific site of release. Capsule coating has been reported as an industrially viable process (2).
rEFERENCES
Colon-specific drug delivery: new approaches and in vitro/in vivo evaluation. Int J Pharm, 235:1-15, 2002.
Cole, E.T., Scott, R.A., Connor, A.L., Wilding, I.R., Petereit, H.U., Schminke, C., Beckert, T. and Cadé, D., Enteric coated HPMC capsules designed to achieve intestinal targeting. Int J Pharm, 231:83-95, 2002.
Khan, A.K.A., Piris, J. and Truelove, S.C., An experiment to determine the active therapeutic moiety of sulphasalazine. Lancet, 2:892-895, 1977.
Peppercorn, M.A., Sulfasalazine: pharmacology, clinical use, toxicity and related new drug development. Ann Intern Med, 3:377-386, 1984.
Zuckner, J., International experience with diclofenac in rheumatoid arthritis. Am J Med Suppl 4B:3942, 1986.
Kendall, M.J., Thornhill, D.P. and Willis, J.V., Factors affecting the pharmacokinetics of diclofenac sodium. Rheumatol Rehabilitation Supp,. 2:3845, 1979.
Willis, J.V., Kendall, M.J., Flinn, R.M., Thornhill, D.P. and Welling, P.G.,. The pharmacokinetics of diclofenac sodium following intravenous and oral administration. Eur J Clin Pharmacol, 16:405410, 1979.
Willis, J.V., Kendall, M.J. and Jack, D.B., The influence of food on the absorption of diclofenac after single and multiple oral doses. Eur J Clin Pharmacol, 19:3337, 1981.
Gleiter, C.H., Antonin, K.H., Bieck, P., Godbillon, J., Schönleber W. and Malchow, H., Colonoscopy in the investigation of drug absorption in healthy volunteers. Gastrointest Endosc, 31: 7173, 1985.
Lorenzo-Lamosa, M.L., Remuñán-López, C., Vila-Jato, J.L. and Alonso, M.J., Design of microencapsulated chitosan microspheres for colonic drug delivery. J Control Rel, 52:109-118, 1998.
Orienti, I., Cerchiara, T., Luppi, B., Bigucci, F., Zuccari, G. and Zecchi, V., Influence of different chitosan salts on the release of sodium diclofenac in colon-specific delivery. Int J Pharm, 238:51-59, 2002.
Singla, A.K. and Chawla, M., Chitosan: some pharmaceutical and biological aspects-an update. J Pharm Pharmacol, 53:1047-1067, 2001.
Sabnis, S., Rege, P. and Block, L.H., Use of chitosan in compressed tablets of diclofenac sodium: inhibition of drug release in an acidic environment. Pharm Dev Technol, 2:243-255, 1997.
Haberlin, B. and Friend, D.R., Anatomy and Physiology of the Gastrointestinal Tract: Implication for Colonic Drug Delivery, in: Friend DR (ed), Oral Colon-Specific Drug Delivery, CRC Press, Florida, pp. 1-43, 1992.
Khalid, M.N., Ho, L., Agnely, F., Grossiord, J.L. and Couarraze, G., Swelling properties and mechanical characterization of a semi-interpenetrating chitosan/polyethylene oxide network. Comparison with a chitosan reference gel. S.T.P. Pharma Sci, 9:359-364, 1999.
Di Colo, G, Zambito, Y, Burgalassi, S, Serafini, A and Saettone, MF, Effect of chitosan on in vitro release and ocular delivery of ofloxacin from erodible inserts based on poly(ethylene oxide). Int J Pharm, 248:115-122, 2002.
Carelli, V., Di Colo, G., Nannipieri, E., Poli, B. and Serafini, M.F., Polyoxyethylene poly(methacrylic acid-co-methyl methacrylate) compounds for site-specific peroral delivery. Int J Pharm, 202:103-112, 2000.
Di Colo, G., Falchi, S. and Zambito, Y., In vitro evaluation of a system for pH-controlled peroral delivery of metformin. J Control Rel, 80:119-128, 2002.
Tozaki, H., Komoike, J., Tada, C., Maruyama, T., Terabe, A., Suzuki, T., Yamamoto, A. and Muranishi, S., Chitosan capsules for colon-specific drug delivery: improvement of insulin absorption from the rat colon. J Pharm Sci, 86: 1016-1021, 1997.
Flynn, G.L., Yalkowsky, S.H. and Roseman, T.J., Mass transport phenomena and models: Theoretical concepts. J Pharm Sci, 63:479-509, 1974.
Park, H.S., Lee, J.Y., Cho, S.H., Baek, H.J. and Lee, S.J, Colon delivery of prednisolone based on chitosan coated polysaccharide tablets. Arch Pharm Res, 25:964-968, 2002.
Corresponding Author: Giacomo Di Colo, Department of Bioorganic Chemistry and Biopharmaceutics, University of Pisa, Via Bonanno 33, 56126 Pisa, Italy. giadic@farm.unipi.it
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps