J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(2):198-204, 2003
Correlation of photosensitizer delivery to lipoproteins and efficacy in tumor and arthritis mouse models; comparison of lipid-based and Pluronic® P123 formulations.
Rubinah K Chowdhary, Isha Sharif, Namrata Chansarkar, David Dolphin1
Department of Chemistry, University of British Columbia, Vancouver, British Columbia, CanadaLeslie Ratkay, Sean Delaney
QLT Inc., Vancouver, British Columbia, CanadaHoward Meadows
Department of Microbiology and Immunology, University of British Columbia, Vancouver, British Columbia, CanadaReceived 8 January 2003, Revised 22 April 2003, Accepted 22 April 2003
PDF version
Abstract
PURPOSE: The purpose of this study was the use of animal models to demonstrate the importance of drug delivery (verteporfin) to plasma lipopropteins in order to attain efficacy of photodynamic therapy (PDT) in vivo. METHODS. Photosensitzers appropriately formulated in various vehicles such as pluronics and lipid-based systems were compared to delivery of the drug in DMSO in two in vivo systems. The first was a tumor model using male DBA/2 mice inoculated intradermally with M1 rhabdomyosarcoma cells and in the second, arthritis in the MRL -lpr mouse strain was enhanced by two intradermal injections of complete Freunds adjunct. RESULTS. Those formulations in which the drug was in a monomeric form were better able to transfer drug to lipoproteins, which in turn led to superior PDT in vivo . CONCLUSIONS. The ability to introduce drug in monomeric form into the circulation correlates well with efficacy of photosensitizer formulations in mouse arthritis and tumor models.
Introduction
The scope of photodynamic therapy (PDT) has recently expanded well the beyond treatment of malignant tumors to conditions with hypervascularization and inflammatory involvement. This includes ocular disorders such as macular degeneration, and a wide range of inflammatory and autoimmune conditions. The majority of photosensitizers of interest for PDT are hydrophobic in nature. When such drugs are injected, they tend to be transported in the bloodstream predominantly associated to lipoproteins (1). Cellular internalization of these drug-lipoprotein complexes via low-density (LDL) receptors, which are upregulated in rapidly dividing tissues undergoing rapid growth or repair, is believed to confer a degree of specificity on the preferential localization of such drugs in diseased tissues. Pre-association of such drugs with LDLs has been shown to result in better delivery to target tissues and improved efficacy in vivo (2).
The tendency of highly hydrophobic photosensitizing drugs to undergo aggregation, in contact with aqueous systems, has a deleterious effect on the photosensitized oxidative process (3). Hence, such drugs need to be introduced into the bloodstream in a form that is either monomeric, or easily dissociated in contact with plasma. For intravenous administration, therefore, hydrophobic photosensitizers require formulation to minimize aggregation in vivo. Verteporfin (benzoporphyrin derivative monoacid ring A, BPDMA 1; Figure 1), which has recently been approved by the FDA for the treatment of macular degeneration, is prepared in a lipid-based formulation to minimize aggregation.
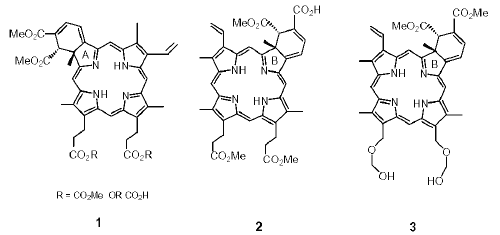
Figure 1: Structures of BPDMA, 1, and ring B derivatives 2 and 3.
The synthetic route for BPDMA results in the production of A- and B-ring intermediates in almost equimolar quantities. Drugs derived from both these intermediates display similar efficacy to BPDMA in various biological test systems. However, the B-ring derivatives of benzoporphyrin have proved to be particularly prone to aggregation (4), and as such have proved considerably more difficult to formulate than their A-ring counterparts. While A-ring compounds are readily prepared in lipid-based formulations, such formulations of the B-ring compounds proved to be unstable, with aggregation taking place within the lipid phase on prolonged storage. This resulted in failure of formulations in biological systems, as well as drug loading which was too low for lipid-based formulations to be clinically viable. Similarly, drug loading in homo-polymeric formulations was also inadequate, but these compounds were successfully formulated in block copolymers known as poloxamers or Pluronics (9). Pluronics are a family of symmetrical triblock copolymers composed of polyethylene oxide (PEO) and polypropylene oxide (PPO), with the form (EO)a -(PO)b -(EO)a (5). Polypropylene oxide (PPO) forms the central hydrophobic core in which the methyl groups are believed to interact through Van der Waals forces with the substance to be solubilized. The PEO block confers water solubility to the polymer through hydrogen bonding interactions of the ether oxygen with water molecules. Poloxamers are readily soluble in aqueous, polar and non-polar organic solvents, and lend themselves well to existing formulation techniques.
Poloxamers have an extensive range of applications in the pharmaceutical industry such as emulsification, solubilization, dispersion, and as thickening, coating and wetting agents (6). Poloxamers have also been used as emulsion stabilizers in intravenous clinical preparations, particularly as emulsion stabilizers in nutrient lipid emulsions and in fluorochemical emulsions developed as oxygen carriers for blood replacement (7).
After extensive screening with various commercially available Pluronics, one of these, Pluronic® P123 (poloxamer 403), or (EO)21 -(PO)67 -(EO)21' was found to be especially useful for formulation a wide range of tetrapyrrolic drugs, including A- and B-ring benzoporphyrin derivatives (8). The reason behind their ability to stabilize drug in monomeric form is presumed to be their ability to form stable micelles (9). Pluronic formulations also displayed superior efficacy in biological systems compared to other carriers. This study investigates their interaction with blood components.
Materials and Methods
Pluronic formulation. Samples of Pluronics were kindly provided by BASF, Mount Olive, N.J., and photosensitizers used in this study were synthesized by Dr E. Sternberg (Chemistry, University of British Columbia). BPDMA was formulated in Pluronic® F127. B-ring compounds 2 and 3, were prepared in P123 due to insufficient drug loading in F127. Drugs were incorporated into Pluronics using the thin film method: drug (1 mg/mL) and Pluronic (5% w/v) were co dissolved in dichloromethane in a round bottom flask. The solvent is then removed by rotary evacuation (Rotavapor R-124, Buchi B171 Vacobox pump), which deposits the Pluronic-drug complex in the form of a thin film, with a high surface area, on the inner surface of the flask. The film was then hydrated at room temperature using an iso-osmolar solution of 9.5% w/v lactose to give the injectable aqueous suspension with a final drug concentration of 2 mg/mL in 10% w/v Pluronic® P123 (100 mg/mL). Following hydration, formulations were kept overnight to allow any slow drug aggregation processes to take place. These aggregates were removed by centrifugation in an Eppendorff bench top microfuge (14,000 g, 30 min), and the supernatant filter sterilized by passing through 0.2mfilters (Millipore, Bedford, MA.) prior to use in animals. Drug concentration in the formulation was determined by using absorbance at 690 nm in methanol. The molar extinction coefficients (ε 690) used for BPDMA (1) and compounds 2 and 3 were 34500 M-1 cm-1, 30425 M-1cm-1 and 31633 M-1 cm-1 respectively.
Lipid-based formulations . These formulations were prepared using the thin film procedure as above; the lipids were DMPC and EPG (Northern Lipids, Vancouver, Canada). A thin film containing the lipids and drug was prepared using dichoromethane. It was hydrated using 9.5% lactose solution to give large multilamellar vesicles. Size reduction was carried out by extrusion (Extruder model 4T, Biomembranes Inc., Vancouver, Canada) through 0.4 micron followed by 0.2-micron pore size polycarbonate filters (5 times through each) at 45°C. Absorbance was used to determine drug concentration as above.
DMSO formulation . Drug was directly dissolved in DMSO and the solution combined with mouse plasma prior to injection into animals.
Drug association with plasma components . The assay for centrifugal separation of plasma components was based on Rudel et al . (10). It was subsequently modified by Alison et al . (1), and was scaled down for this study. Fresh human plasma was collected in EDTA tubes (average n=3), and KBr added to give a concentration of 1.21-1.23 g/mL. Photosensitizer formulations were added to 0.8 mL pre-warmed plasma (37°C) to give a final concentration of 10 m g/mL. Plasma was under layered with 2.45 mL KBr/EDTA at 1.21 g/mL in polycarbonate tubes. Samples were centrifuged at 512K g (100,000 RPM, Beckman TLA 100.3 rotor) for 16-18 h at 20°C. Layer positions were marked to allow determination of layer volume. Each layer was sampled by removing a portion using a syringe inserted from the top. Known volumes of plasma layers were removed into 1% Triton X-100 in PBS in an 1.8 mL tube (Eppendorf Scientific Inc., Westbury, NY.). Samples were vortex mixed and then centrifuged for 2 min at 14,000 RPM in an Eppendorf centrifuge for clarification. Fluorescence at 690 nm (λex=434 nm) was read alongside standards of known drug concentration. Total drug present in each layer was calculated based on known layer volume and fluorescence values. The validity of this assay was tested using MACE (monoaspartyl chlorin e6), a relatively water-soluble photosensitizer known to be bound and transported by albumin in the circulation. MACE was found to be predominantly associated with albumin (87%), with very little in the lipoprotein layer (11%). BPDMA, which is known to be associated with the lipoproteins fraction was used as a control.
Comparison of the performance of drug formulations in a mouse tumor model. The tumor model used was the DBA/2 mouse (male) inoculated intradermally with M1 rhabdomyosarcoma tumor cells (M1, ATCC). When tumors reached a diameter of 4-6 mm, the mice (n=10) were weighed and treated with PDT, which involved intravenous injection of the appropriate dose of formulated drug in a volume of 0.2 mL 5% dextrose. Fifteen minutes later, the tumor region was exposed to laser light (Argon pumped dye laser (5W), 690nm, 50J/cm2). Animals were then monitored for tumor recurrence over a 20-day period post treatment.
Comparison of drug formulations in a mouse arthritis model. Arthritis in the MRL -lpr mouse strain was enhanced by giving 2 intradermal injections (thoracic and inguinal sites) with 0.05 mL of complete Freunds adjuvant (CFA) containing 10 mg/mL heat-inactivated M. Tuberculosis . PDT was given on days 0, 10 and 20 following CFA treatment. Ankle width measurements were taken every 5 days prior to PDT treatment. PDT was carried out as follows; MRL-lpr mice (average n= 15) were injected intravenously at 0.5 mg/kg (copolymer or liposomal formulations). One group was injected with copolymer alone at an equivalent copolymer concentration to that found in the formulation. Animals were protected from light until they were exposed to light at 80 J/cm2 for 1.5 h (8 mW/cm2 ) an hour later using a light box with a tungsten-halogen light filtered between 560-900 nm. Light intensity was measured using an 1L 1350 photometer, International Light Inc., Newburgport, MA.
RESULTS AND DISCUSSION
Plasma Distribution of Photosensitizers Delivered by Block Copolymer and Lipid-based Formulations . Tables 1-3 show how different formulations of BPDMA and B-ring photosensitizers affect their pattern of distribution among components of fractionated plasma.
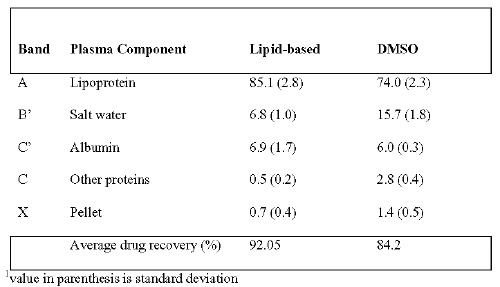
Table 1: Percent association of BPDMA (1) with various fractions following centrifugal separation of plasma1
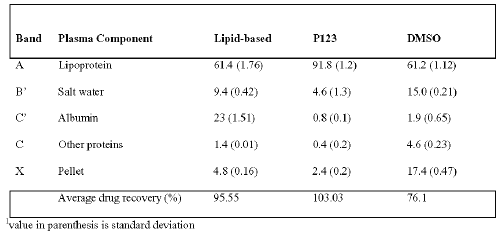
Table 2: Percent association of B-ring compound 2 with various fractions following centrifugal separation of plasma1.
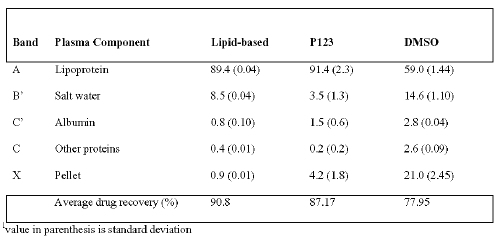
Table 3: Percent association of B-ring compound 3 with various fractions following centrifugal separation of plasma1.
Lipid-based BPDMA associated predominantly with the lipoproteins when mixed with plasma, and this confirms observations in previous reports [1]. The Pluronic formulation of B-ring Compound 2 was found to be more effective at delivering it to the lipoprotein fraction in comparison its lipid-based formulation (Table 2). In the case of B-ring compound 3, delivery to lipoproteins was comparable to the lipid-based formulation. Association of drug with lipoproteins on mixing different formulations with plasma correlates well with the therapeutic efficacy of PDT in animal models. The Pluronic formulation of B-ring compound 2 outperforms the lipid-based one, whereas the lipid-base formulation of B-ring compound 3 also demonstrates good potency in vivo.
DMSO solutions, particularly of the B-ring compounds tended to deliver drug poorly to the lipoproteins. Although BPDMA is the least likely out of the three drugs tested to aggregate, Table 1 shows that addition of BPDMA/DMSO to plasma results in inefficient delivery of the drug to the lipoprotein fraction in comparison to the lipid-based formulation. All drugs added to plasma in DMSO resulted in high ( ~ 15%) drug concentration in the salt/water fraction, which could reflect dimers, or small aggregates of drug, possibly in the form of micelles. Similarly, the larger proportion of DMSO-delivered drug in the pellet could reflect larger aggregates, and this is particularly evident in the B-ring compounds. Low total drug recoveries were observed in DMSO formulations of all three drugs, which probably reflect difficulty in fluorimetric sampling of the pelleted drug due to excessive aggregation. Aggregation can drastically reduce the fluorescence yield of tetrapyrrolic drugs; fluorescence studies (results not shown) indicate that aggregation can be present at very low concentrations of BPDMA, and dissociation does not take place readily even in the presence of whole plasma. On the other hand, in Pluronic formulations, even B-ring drugs are known to be stabilized in monomeric form. Fluorescence yield in Pluronics is high and fluorescence is not concentration quenched; extinction coefficients of drugs in aqueous Pluronic suspensions are similar to those in organic solvents such as dichloromethane, which are known to be good solvents for these drugs.
Apart from altered spectroscopic properties and reduced photosensitizing capacity of aggregated photosensitizers, an additional implication of aggregation could be reduced binding to lipoproteins in the circulation. This can impact negatively on PDT because most target tissues, such as those undergoing rapid proliferation or repair, tend to express high levels of LDL receptors, and lipoprotein mediated delivery could enhance selective accumulation of photosensitizers in these tissues. In vivo , aggregation that is not readily reversible in the circulation could therefore translate into altered biodistribution and loss of selectivity and efficacy of PDT.
The above experiment demonstrates that for hydrophobic drugs, the Pluronic and lipid-based formulations deliver a greater proportion of drug to the lipoprotein fraction. In the case of compound 2, there is considerable difference between Pluronic and liposomal delivery of drug to the lipoprotein fraction, with a surprisingly high proportion of the lipid phase delivered drug bound to the albumin fraction. Although albumin is the most abundant protein in circulation, the number of binding sites it has for tetrapyrrolic drugs is limited. It has been observed that binding of certain porphyrins, such as TPPS4 (meso-tetrakis(p-sulphonatophenyl) porphyrin) with albumin promotes aggregation to take place at the proteins surface (11). Lipid-based and Pluronic formulations of compound 3, on the other hand, display identical delivery to lipoproteins, but with a somewhat higher binding of liposomal drug to the albumin fraction.
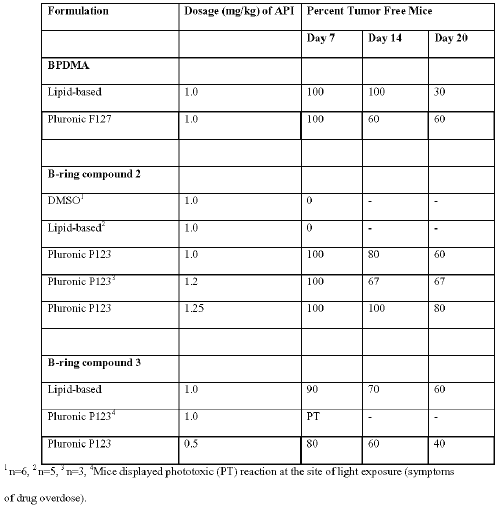
Comparison of tumor recurrence in mice model treated with PDT using A- & B- ring photosensitizers in block copolymer and lipid-based formulations. Table 4 summarizes the performance of BPDMA and different formulations of B-ring compounds. The lipid-based BPDMA formulation was used as a standard for assessing performance of other photosensitizers and formulations. It was observed with BPDMA that at the end of the 20-day period, mice treated with the Pluronic formulation were twice as likely to remain tumor free compared to those treated with lipid-based BPDMA.
Table 4: Tumor Cure Following PDT using different formulations of BPDMA and B-ring photosensitizers.
This could reflect improved photosensitizer binding to lipoproteins upon intravenous injection, and better lipid phase mediated delivery to target tissues by the Pluronic formulated BPDMA. Additionally, enhanced availability resulting from the ability of Pluronics to enhance membrane permeability and promote drug transfer across the plasma membrane could also be a contributing factor (12). On comparing the performance of lipid-based formulations of the three drugs in the mouse tumor model, compound 3 demonstrated the greatest potency in spite of not being suitable for lipid-based formulations in terms of drug loading (requires very high lipid:drug). Administration of 1 mg/kg compound 3, formulated in P123, to tumor bearing mice resulted in a strong phototoxic reaction (edema) at the site of exposure immediately following irradiation, and the animals had to be euthanized. This observation is typical of overdosing of the photosensitizer, and suggested that more effective drug delivery is achievable using Pluronics compared to the lipid-based formulations at the same drug dosage. At a lower dose of 0.5 mg/kg (API) delivered in Pluronic, a cure rate was achieved similar to that of lipid-based formulations of compound 3 and BPDMA at 1 mg/kg.
The performance of compound 2 in the mouse tumor model demonstrated greatest sensitivity to the formulation agent used. At 1 mg/kg, the plasma-DMSO preparation was found to be completely ineffective for PDT purposes. Similarly, the lipid-based formulation of compound 2 was ineffective in vivo . This correlates well with inadequate delivery of drug to plasma lipoproteins by both DMSO and lipid-based preparations of compound 2. The performance of compound 2 formulated in Pluronic at 1 mg/mL was marginally better than that of lipid-based BPDMA, and comparable to BPDMA in Pluronic. Increasing the dose of the Pluronic formulation of compound 2 by 25% resulted in a marked dose dependent improvement in performance in the tumor assay.
It has been shown that low drug delivery to lipoproteins ( ~60%), as in the case of DMSO formulations of both B-ring drugs and lipid-based compound 2, results in complete failure in the tumor assay. This observation suggests a strong role for lipoprotein delivery of drugs to the target tissues, particularly in the case of highly hydrophobic photosensitizers.
Comparison of copolymer and lipid-based formulations of compound 2 and 3 for PDT treatment of arthritis in MRL-lpr mouse model. This experiment evaluated the ability of various drug formulations to control ankle inflammation in mice using the protocol described for liposomal BPDMA (13). While it is unclear how PDT alleviates arthritis (14) we suggest that the activated cells, responsible for maintaining inflammation, are selectively targeted and then modified by PDT. It has been shown previously that activated T lumphocytes display greater sensitivity to PDT than resting cells (15). The results shown in Figure 2 suggest that as for tumor control, lipid-based compound 3 was most effective at controlling inflammation. Pluronic formulations of 2 and 3 were also equally successful in preventing the development of CFA-enhanced arthritis in mice. The formulation that displayed almost complete failure in this model, performing only marginally better than the drug-free Pluronic control, was lipid-based compound 2. This formulation also failed in the tumor model, and failed to deliver adequate drug to the lipoprotein fraction of plasma.
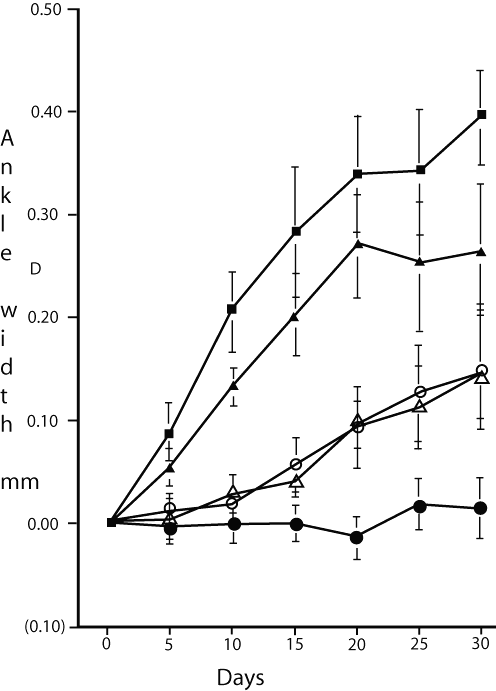
Figure 2: Inflammation control measured by ankle width following PDT treatments at days 0, 10 and 20 following enhancement of arthritis in the MRL -lpr mouse model. ----●---- Lipid based compound 3; ----▲---- lipid based compound 2; ----m---- Pluronic based compound 3; ----Δ---- Pluronic based compound 2; ----■---- Pluronic only. All measurements were made blind.
Interestingly, with the exception of compound 3 in the arthritis model, Pluronic formulations displayed better performance in vivo. Delivery of 3, in Pluronic formulation, to plasma lipoproteins was similar to the lipid-based formulation, as was efficacy in the tumor model. Different tissue characteristics and cellular involvement in the two disease models could account for the difference in performance of the two formulations in vivo. It is also possible that the nature of the interaction of the drug with the carrier system, and that of the drug-carrier complex with lipoproteins could also play a role. Drug-lipid interaction in the lipid phase has been shown to be highly specific; and whereas release characteristics of 2 are similar to those of BPDMA from the same lipid phase system, release of lipid-based compound to serum did not take place as readily (8). Drug-lipid interaction could greatly influence behavior of the lipid phase in vivo, e.g. by altering fluidity, the half-life of the lipid phase in the circulation and the mode of transfer of drug to lipoproteins. The relative affinities of the drug for the delivery vehicle, lipoproteins and plasma membranes could also affect the mechanism of drug uptake by cells and tissues.
Efficacy of PDT in vivo depends on numerous additional factors such as extent and condition of vasculature in the target tissue. Continuity of endothelium is disrupted in certain types of tumors and this allows better perfusion of plasma through the tissues, and in the case of particulate delivery systems, prolonged and higher drug retention relative to healthy tissues. Uptake on a cellular level by diseased tissue or microvascular endothelium within the target tissue is also an important factor, whether LDL-mediated or by pinocytosis. Equally important is intracellular retention of the drug especially at the time of light exposure. Destruction of the vascular system is an important determinant of tissue ablation using PDT, and is generally believed to be dependent on high plasma levels of circulating drug, particularly in the case of water-soluble photosensitizers, which do not readily traverse plasma membranes into cells. In the case of hydrophobic benzoporphyrin-based derivatives, plasma levels of the drug drop rapidly after infusion, presumably due to rapid tissue uptake by endothelial cells at the front line of vascular delivery of photosensitizers. The vascular effect in the case of hydrophobic drugs could therefore be very potent, especially at earlier irradiation times. Cellular uptake and biodistribution studies are reported elsewhere (8).
Drug delivery and performance in vivo is therefore a complex area of study. Our studies suggest that efficient drug delivery to lipoproteins is an important determinant of drug potency in vivo. Whether drug uptake is in fact lipoprotein receptor mediated could be determined by competitive uptake studies in cell culture, either using antibodies against the LDL receptor or induction of high LDL receptor expression by nutrient deprivation. Direct partitioning of drugs from lipoproteins in the circulation directly to plasma membranes is also a possibility for highly hydrophobic drugs. Transfer of BPDMA between formulation agent and biochemical entities with hydrophobic sites with drug binding capacity has been demonstrated. Drug exchange and transfer of lipid-based drug to plasma components such as tryptophan and lipoproteins has been shown to take place readily (16).
The nature of the delivery vehicle is of utmost importance as it determines the nature of interaction with plasma components. In delivery using lipid-based and micellar Pluronic systems, the drug is better protected from exposure to the aqueous environment of the circulation. Drug in DMSO exhibits rapid aggregation on exposure to aqueous systems, and, as this aggregation is not readily reversible in plasma, efficacy of PDT is vastly reduced. In conclusion, deliveries of photosensitizers in systems that minimize aggregation provide better partitioning into the lipoprotein fraction, as well as good performance in vivo .
ACKNOWLEDGMENTS
We gratefully acknowledge financial support from NSERC in the form of a CRD grant for work carried out jointly between the Department of Chemistry, UBC and QLT Inc., Vancouver, B.C.
References
Allison B.A, Pritchard, P.H., Richter, A.M., and Levy, J.G., The plasma distribution of benzoporphyrin derivative and the effects of plasma lipoproteins on its biodistribution. Photochem. Photobiol. 52:501-507, 1990.
Allison B. A., Waterfield, D., Richter, A. M. and Levy J. G., The effect of plasma lipoproteins on in vitro tumor cell killing and in vivo tumor photosensitization with benzoporphyrin derivative. Photochem. Photobiol. 54:709-715 (1991).
Aveline B. M., Hasan, T. and Redmond, R. W., The effects of aggregation, protein binding and cellular incorporation on the photophysical properties of benzoporphyrin derivative monoacid ring A (BPDMA). J Photochem. Photobiol. B30:161-169, 1995.
Delmarre D., Hioka, N.; Boch, R.; Sternberg, E. and Dolphin, D., Aggregation studies of benzoporphyrin derivative. Can. J. Chem. 79:1068, 2001.
Schmolka I. R., A review of block polymer surfactants. J. Am. Oil Chemists Soc. 54:110, 1977.
Schmolka I. R., Poloxamers in the pharmaceutical industry in Polymers for controlled drug delivery. Ed P. J. Tarcha. CRC Press Boca Raton, pp 189-214, 1991.
Mitsuno T., Ohyanagi, H. and Yokayama, K., Development of perfluorochemical emulsions as blood gas carrier. Artif. Organs, 8:25, 1984.
Chowdhary R. K., N. Chansarkar, N., Sharif, I., Hioka, N. and Dolphin, D., Formulation of chlorin-based photosensitizers in Pluronics. Photochem. Photobiol. 77:229 2003.
9. Alexandridis P. and Hatton, T.A., Poly(ethylene oxide)-poly(propylene oxide) block copolymer surfactants in aqueous solutions and at interfaces: thermodynamics, structure, dynamics, dynamics and modelling. Colloids and surfaces A: Physicochemical and Engineering Aspects, 96:1-46, 1995.
Rudel LL., Lee, J.A., Morris, M.D. and Felts, J.M., Characterization of plasma lipoproteins separated and purified by agarose-column chromatography. Biochem J. 139:89-95, 1974.
Yu S., Egorov, A.A., Savitsky, A.P. and Papkovsky, D.B., Bull. Exp. Biol. Med. 109:349-351, 1990.
Batrakova E.V., Han, H.Y., Miller, D.W. and Kabanov, A.V., Effects of Pluronic P85 unimers and micelles on drug permeability in polarized BBMEC and Caco-2 cells. Pharm. Res. 15:1525-1532, 1998.
Chowdhary R. K., Ratkay, L., Neyndorff, H.C., Richter, A., Obochi, M., King, D., Jain, A., Waterfield, J.D. and Levy, J.G., The use of transcutaneous photodynamic therapy in the prevention of adjuvant enhanced arthritis in MRL-lpr mice. Clin. Immunol. Immunopathol., 72:255-263, 1994.
Chowdhary R. K., Ratkay, L., Canaan, A.J., Waterfield, J.D., Richter, A.M., and Levy, J.G., Uptake of Verteporfin by articular tissues following systemic and intra-articular administration. Biopharm. & Drug Disposition, 19:395, 1998.
Hunt, D.W.C., Jiang, H., Granville, D.J., Chan, A.H., Leong, S., and Levy, J.G., Consequences of the photodynamic treatment of resting and activated peripheral T lymphocytes. Immunopharmacology, 41:31, 1999.
Chowdhary R. K., Shariff, I. and Dolphin, D., Drug release characteristics of lipid based benzoporphyrin derivatives. J. Pharm. & Pharmaceutical Sciences, 6:13, 2003.
Corresponding Author: David Dolphin, Department of Chemistry, 2036 Main Mall, University of British Columbia, Vancouver, British Columbia, Canada, V6T 1Z1. ddolphin@qltinc.com
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps