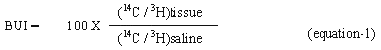J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(2):252-273, 2003
Drug delivery to the central nervous system: a review.
Ambikanandan Misra1, Ganesh S., Aliasgar Shahiwala
Pharmacy Department, M.S. University of BarodaShrenik P. Shah
Sun Pharma Advanced Research Center, BarodaReceived 16 June 2003, Revised 26 June 2003, Accepted 05 August 2003
PDF version
Abstract
The brain is a delicate organ, and evolution built very efficient ways to protect it. Unfortunately, the same mechanisms that protect it against intrusive chemicals can also frustrate therapeutic interventions. Many existing pharmaceuticals are rendered ineffective in the treatment of cerebral diseases due to our inability to effectively deliver and sustain them within the brain. General methods that can enhance drug delivery to the brain are, therefore, of great interest. Despite aggressive research, patients suffering from fatal and/or debilitating central nervous system (CNS) diseases, such as brain tumors, HIV encephalopathy, epilepsy, cerebrovascular diseases and neurodegenerative disorders, far outnumber those dying of all types of systemic cancer or heart disease. The clinical failure of much potentially effective therapeutics is often not due to a lack of drug potency but rather to shortcomings in the method by which the drug is delivered. Treating CNS diseases is particularly challenging because a variety of formidable obstacles often impede drug delivery to the brain and spinal cord. By localizing drugs at their desired site of action one can reduce toxicity and increase treatment efficiency. In response to the insufficiency in conventional delivery mechanisms, aggressive research efforts have recently focused on the development of new strategies to more effectively deliver drug molecules to the CNS. This review intends to detail the recent advances in the field of brain-targeting, rational drug design approach and drug delivery to CNS. To illustrate the complexity of the problems that have to be overcome for successful brain targeting, a brief intercellular characterization of the blood-brain barrier (BBB) is also included.
Introduction
Despite enormous advances in brain research, brain and central nervous system disorders remain the world's leading cause of disability, and account for more hospitalizations and prolonged care than almost all other diseases combined. The major problem in drug delivery to brain is the presence of the BBB. Drugs that are effective against diseases in the CNS and reach the brain via the blood compartment must pass the BBB. In order to develop drugs which penetrate the BBB well to exhibit the expected CNS therapeutic effects, it is of great importance to understand the mechanisms involved in uptake into and efflux from the brain. The function of the BBB is dynamically regulated by various cells present at the level of the BBB (1). This realization implies better understanding of the relationship of transport at the BBB to drug structure and physicochemical properties.
Despite successful examples of drug delivery to the CNS, but only some have reached the phase where they can provide safe and effective human applications. As pharmacological strategies improve, there will be less need for invasive procedures for treating CNS diseases. Considerable strides have been made in intravascular delivery and neurosurgical invasive procedures to deliver therapeutic substances into the brain.
This review will prove invaluable to researchers interested in the fundamental function of the BBB and those in the pharmaceutical industry interested in rational drug design directed at delivering drugs to the brain.
Barriers to CNS drug delivery
The failure of systemically delivered drugs to effectively treat many CNS diseases can be rationalized by considering a number of barriers that inhibit drug delivery to the CNS.
Blood-Brain Barrier
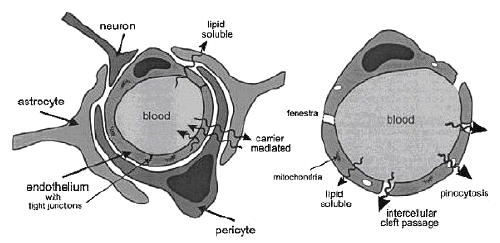
It is now well established that the BBB is a unique membranous barrier that tightly segregates the brain from the circulating blood (2, 3). The CNS consist blood capillaries which are structurally different from the blood capillaries in other tissues; these structural differences result in a permeability barrier between the blood within brain capillaries and the extracellular fluid in brain tissue. Capillaries of the vertebrate brain and spinal cord lack the small pores that allow rapid movement of solutes from circulation into other organs; these capillaries are lined with a layer of special endothelial cells that lack fenestrations and are sealed with tight junctions. Tight epithelium, similar in nature to this barrier, is also found in other organs (skin, bladder, colon, and lung) (4).This permeability barrier, comprising, the brain capillary endothelium, is known as the BBB. Ependymal cells lining the cerebral ventricles and glial cells are of three types. Astrocytes form the structural frame work for the neurons and control their biochemical environment. Astrocytes foot processes or limbs that spread out and abutting one other, encapsulate the capillaries are closely associated with the blood vessels to form the BBB. Oligodendrocytes are responsible for the formation and maintenance of the myelin sheath, which surrounds axons and is essential for the fast transmission of action potentials by salutatory conduction. Microglias are blood derived mononuclear macrophages. The tight junctions between endothelial cells results in a very high trans-endothelial electrical resistance of 1500-2000 W.cm2 compared to 3-33 W.cm2 of other tissues which reduces the aqueous based para-cellular diffusion that is observed in other organs (5, 6).
Micro-vessels make up an estimated 95% of the total surface area of the BBB, and represent the principal route by which chemicals enter the brain. Vessels in brain were found to have somewhat smaller diameter and thinner wall than vessels in other organs. Also, the mitochondrial density in brain micro-vessels was found to be higher than in other capillaries not because of more numerous or larger mitochondria, but because of the small dimensions of the brain micro-vessels and consequently, smaller cytoplasmic area. In brain capillaries, intercellular cleft, pinocytosis, and fenestrae are virtually nonexistent; exchange must pass trans-cellularly. Therefore, only lipid-soluble solutes that can freely diffuse through the capillary endothelial membrane may passively cross the BBB. In capillaries of other parts of the body, such exchange is overshadowed by other nonspecific exchanges. Despite the estimated total length of 650km and total surface area of 12 m2 of capillaries in human brain, this barrier is very efficient and makes the brain practically inaccessible for lipid- insoluble compounds such as polar molecules and small ions. As a consequence, the therapeutic value of many promising drugs is diminished, and cerebral diseases have proved to be most refractory to therapeutic interventions. Given the prevalence of brain diseases alone, this is a considerable problem. Practically all drugs currently used for disorders of the brain are lipid-soluble and can readily cross the BBB following oral administration. Although antimicrobial b-lactam antibiotics, when administered intracerebroventricularly, cause severe convulsion, fortunately these antibiotics, when administered intravenously or orally, do not cause such central nervous system (CNS) side effect because their limited transport across the blood-brain barrier (BBB). Further, in spite of being well distributed into various tissues, a lipophilic new quinolone antimicrobial agent, grepafloxacin, cannot enter the brain, resulting in the avoidance of CNS side effects such as headache and dizziness due to the displacement of g-aminobutyric acid (GABA) from the GABA receptor binding sites. On the other hand, benzodiazepines such as diazepam have been used as sedative-hypnotic agents, because these lipophilic drugs readily cross the BBB. However, the BBB transport of an immunosuppressive agent, cyclosporin A, which is more lipophilic than diazepam, is highly restricted. Similarly, almost all of the lipophilic anticancer agents such as doxorubicin, epipodophylotoxin and Vinca alkaloids (e.g., vincristine and vinblastine) hardly enter the brain, causing difficulty in the treatment of brain tumors. Although levodopa, which is useful for treatment of Parkinson's disease, is very hydrophilic, it can readily penetrate the BBB. What mechanisms underlie these diverse BBB transport characteristics of drugs which are apparently structurally and pharmacologically unrelated? In order to avoid overlap with this section, the drug transport across the BBB of small-molecular drugs by carrier-mediated transport and of peptide drugs by the adsorptive-mediated transcytosis are discussed in section 7.1.4 and 7.1.5 respectively.
Some regions of the CNS do not express the classical BBB capillary endothelial cells, but have micro-vessels similar to those of the periphery. These areas are adjacent to the ventricles of the brain and are termed the circumventricular organs (CVOs). The CVOs include the choroid plexus, the median eminence, neurohypophysis, pineal gland, organum vasculosum of the lamina terminalis, subfornical organ, subcommisaral organ and the area postrema. Though in the CVO brain regions the capillaries are more permeable to solutes, the epithelial cells of the choroid plexus and the tanycytes of other regions form tight junctions to prevent transport from the abluminal extracellular fluid (ECF) to the brain ECF. The choroid plexus may be of importance when considering the transport of peptide drugs, because it is the major site of cerebrospinal-fluid (CSF) production, and both the CSF and brain ECF freely exchange (7).
The BBB also has an additional enzymatic aspect. Solutes crossing the cell membrane are subsequently exposed to degrading enzymes present in large numbers inside the endothelial cells that contain large densities of mitochondria, metabolically highly active organelles. BBB enzymes also recognize and rapidly degrade most peptides, including naturally occurring neuropeptides (8, 9).
Finally, the BBB is further reinforced by a high concentration of P-glycoprotein (Pgp), active -drug-efflux-transporter protein in the luminal membranes of the cerebral capillary endothelium. This efflux transporter actively removes a broad range of drug molecules from the endothelial cell cytoplasm before they cross into the brain parenchyma. Figure-1 gives a schematic representation of all these BBB properties using a comparison between brain and general capillaries.
Figure 1: Schematic comparison between general (left) and brain (right) capillaries.
Blood-Cerebrospinal Fluid Barrier
The second barrier that a systemically administered drug encounters before entering the CNS is known as the blood-cerebrospinal fluid barrier (BCB). Since the CSF can exchange molecules with the interstitial fluid of the brain parenchyma, the passage of blood-borne molecules into the CSF is also carefully regulated by the BCB. Physiologically, the BCB is found in the epithelium of the choroids plexus, which are arranged in a manner that limits the passage of molecules and cells into the CSF. The choroid plexus and the arachnoid membrane act together at the barriers between the blood and CSF. On the external surface of the brain the ependymal cells fold over onto themselves to form a double layered structure, which lies between the dura and pia, this is called the arachnoid membrane. Within the double layer is the subarachnoid space, which participates in CSF drainage. Passage of substances from the blood through the arachnoid membrane is prevented by tight junctions (10). The arachnoid membrane is generally impermeable to hydrophilic substances, and its role is forming the Blood-CSF barrier is largely passive. The choroid plexus forms the CSF and actively regulates the concentration of molecules in the CSF. The choroid plexus consist of highly vascularized, "cauliflower-like" masses of pia mater tissue that dip into pockets formed by ependymal cells. The preponderance of choroid plexus is distributed throughout the fourth ventricle near the base of the brain and in the lateral ventricles inside the right and left cerebral hemispheres. The cells of the choroidal epithelium are modified and have epithelial characteristics. These ependymal cells have microvilli on the CSF side, basolateral interdigitations, and abundant mitochondria. The ependymal cells, which line the ventricles, form a continuous sheet around the choroid plexus. While the capillaries of the choroid plexus are fenestrated, non-continuous and have gaps between the capillary endothelial cells allowing the free-movement of small molecules, the adjacent choroidal epithelial cells form tight junctions preventing most macromolecules from effectively passing into the CSF from the blood (11). However, these epithelial-like cells have shown a low resistance as compared the cerebral endothelial cells, approximately 200 W.cm2, between blood and CSF (12).
In addition, the BCB is fortified by an active organic acid transporter system in the choroids plexus capable of driving CSF-borne organic acids into the blood. As a result a variety of therapeutic organic acids such as the antibiotic penicillin, the anti-neoplastic agent methotrexate, and the antiviral agent zidovudine are actively removed from the CSF and therefore inhibited from diffusing into the brain parenchyma. Furthermore, substantial inconsistencies often exist between the composition of the CSF and interstitial fluid of the brain parenchyma, suggesting the presence of what is sometimes called the CSF-brain barrier (13). This barrier is attributed to the insurmountable diffusion distances required for equilibration between the CSF and the brain interstitial fluid. Therefore, entry into the CSF does not guarantee a drug's penetration into the brain.
Blood-Tumor Barrier
Intracranial drug delivery is even more challenging when the target is a CNS tumor. The presence of the BBB in the microvasculature of CNS tumors has clinical consequences. For example, even when primary and secondary systemic tumors respond to chemotherapeutic agents delivered via the cardiovascular system, intracranial metastases often continue to grow. In CNS malignancies where the BBB is significantly compromised, a variety of physiological barriers common to all solid tumors inhibit drug delivery via the cardiovascular system. Drug delivery to neoplastic cells in a solid tumor is compromised by a heterogeneous distribution of microvasculature throughout the tumor interstitial, which leads to spatially inconsistent drug delivery. Furthermore, as a tumor grows large, the vascular surface area decreases, leading to a reduction in trans-vascular exchange of blood-borne molecules. At the same time, intra-capillary distance increases, leading to a greater diffusional requirement for drug delivery to neoplastic cells and due to high interstitial tumor pressure and the associated peri-tumoral edema leads to increase in hydrostatic pressure in the normal brain parenchyma adjacent to the tumor. As a result, the cerebral microvasculature in these tumor adjacent regions of normal brain may be even less permeable to drugs than normal brain endothelium, leading to exceptionally low extra-tumoral interstitial drug concentrations (14). Brain tumors may also disrupt BBB, but these are also local and nonhomogeneous disruptions (15).
In conclusion, the delivery of drugs to the CNS via the cardiovascular system is often precluded by a variety of formidable barriers including the BBB, the BCB, and the BTB.
Efflux mechanisms in drug transport to the brain
A detailed understanding of the uptake and efflux mechanisms at the BBB would be very helpful for targeting drugs to the brain to provide the expected CNS pharmacological effect or for the reduction of BBB penetration of drugs in order to minimize side effects in the CNS. Most in-vivo experimental methods describing drug uptake into brain will automatically incorporate any activity of CNS efflux into their apparent determination of brain penetration. Within the CNS are a number of efflux mechanisms that will influence drug concentrations in the brain. Some of these mechanisms are passive while others are active. Active efflux from the CNS via specific transporters may often reduce the measured penetration of drug at the BBB to levels that are lower than might be predicted from the physicochemical properties of the drug, for example, its lipid solubility. The activity of these efflux mechanisms influence the concentration in brain extracellular fluid of free drugs that are available to interact with drug receptor sites. Recently much attention has been focused on the so-called multi-drug transporters; multi-drug resistance protein (MRP), P-glycoprotein (Pgp) and the multi-specific organic anion transporter (MOAT), which belong to the members of the ABC cassette (ATP-binding cassette) of transport protein (16, 17). The MRP in humans appears to be five isoforms, and there are different levels of expression of these various isoforms in different tissues. Pgp is the product of the multidrug resistance (MDR) gene in humans and accepts a wide range of lipid-soluble substrates and will actively efflux these from cells expressing the gene product. The MOAT in the choroid plexus shows some similarity in its substrate preferences with MRP. Noticeably, brain exposure can be increased not only by enhancing influx, but by restricting efflux through the BBB as well. Hence, strategies directed at increasing brain uptake of drugs that are substrates for specific efflux mechanisms need to be focused on designing reactivity with a transporter out of a drug molecule or by examining ways of inhibiting the activity of an efflux mechanism by co-administering a competitive or noncompetitive inhibitor of the efflux pump together with the desired drug. For example, for certain Pgp substrates, coadministeration of a Pgp inhibitor can increase not only oral absorption, but also BBB permeability (18, 19). Coadministration of the Pgp blocker valspodar has recently been shown to not only increase the brain levels pf paclitaxel, but also to considerably improve its therapeutic effect on tumor volume in mice (20). On the contrary, among the brain drug delivery strategies to be discussed later, chemical drug delivery systems (CDDS) are the only ones attempting to not only increase influx, but also to decrease efflux. This strategy is done by exploiting a sequential metabolic approach that first increases influx by passive diffusion through increased lipophilicity and then decreases efflux by a `lock-in' mechanism.
Physicochemical Factors that influence Brain Uptake
Brain penetration, brain uptake, and ability to cross the BBB need to be defined exactly to understand concepts involved in brain uptake. Hence, the various ways in which transfer across the BBB are defined in table-1.
Table 1: Measures of "Brain Uptake".
Biological activity is a general measure of brain uptake. The hypnotic activity of a number of congeneric series of CNS depressants reached a maximum when log octanol-water partition coefficient (log Po/w) was near to 2. Various researchers confirmed this finding and the "rule of 2" became generally accepted (21). But the difficulty here is that the biological activity will depend on at least two factors:
- rate of transfer from blood to brain, or distribution between blood and brain; and
- interaction between drug and some receptors in the brain.
If these two factors cannot be distinguished, then it is impossible to use biological activity as a measure of either rate or equilibrium transfer.
The log Po/w probably still represents the most informative physicochemical parameter used in medicinal chemistry and countless examples where it proved as useful descriptors are available in the literature (22). On the other hand, increasing lipophilicity with the intent to improve membrane permeability might not only make chemical handling difficult, but also increase the volume of distribution in particular plasma protein binding and tends to affect all other pharmacokinetic parameters (23, 24). Furthermore, increasing lipophilicity tends to increase the rate of oxidative metabolism by cytochromes P450 and other enzymes (23, 25). Hence, to improve bioavailability, the effects of lipophilicity on membrane permeability and first pass metabolism have to be balanced.
The brain uptake index (26) is a more rigorous measure of brain uptake in which there is a relative measure of brain uptake by intra-carotid injection of a mixture of 14C-labeled compound and 3H-labeled water (i.e. a saline solution in 3H-labeled water). The radioactivity in brain tissue is recorded 15 seconds after administration, and a brain uptake index (BUI) is defined in equation-1
where the BUI for water is 100. Although, the BUI is useful as a rank order index of brain uptake, is not easily amenable to analysis by physicochemical methods.
A more well-defined measure of rapid brain uptake is the permeability, expressed either as a permeability-surface area product (PS) or as a permeability coefficient (PC), obtained by intravenous injection and measurement of the drug profile in arterial blood. Both the PS product and PC are quantitative measures of the rate of transport obtained by in-situ vascular perfusion technique (27) and so are amenable to analysis through standard physicochemical procedures. An advantage of the perfusion technique as a measure of brain uptake is that the time scale for determination of PS products is very short, so that back transport and biological degradation are minimized. Although there are numerous physicochemical studies on brain perfusion, it is not possible to reach any general conclusions.
Following systemic drug administration, uptake from the circulation into parenchyma by a specific organ of interest will be determined by the following factors: (a) blood flow to the organ, (b) permeability of the micro-vascular wall, and (c) the amount of drug available for uptake, which is inversely related to systemic clearance and is represented by the area under the plasma concentration-time curve (AUC). For the quantification of brain tissue accumulation (Cbrain) at time T during the phase of unidirectional uptake, the following equation-2 holds:
where PS is the brain capillary permeability surface area product, an expression equivalent to the organ clearance and AUC is the area under the plasma concentration time curve. It should be mentioned that this equation does not take into account efflux of either intact drug or metabolism and efflux of degradation products from the brain. Measurement of efflux is covered in section 6 of this review.
Based on the relationship between the octanol / water partition coefficient (PC) divided by the square root of the molecular weight (PC/ Mw1/2) and the BBB permeability coefficient (PS), one can classify at least three different groups: (a) substrates exhibiting a good correlation, (b) substrates exhibiting a significantly greater PS value than indicated by their lipophilicity, and (c) substrates exhibiting a significantly smaller PS value than indicated by their lipophilicity. The transport mechanism for groups (a) and (b) is passive diffusion and facilitated transport, respectively (27). The molecular weight of the compounds in group (c) is greater than 400 Da., the absolute cut-off for significant BBB passage regardless of lipophilicity. This molecular weight threshold hypothesis was proposed to explain the mechanism operating in the case of group (c) (28).
Brain uptake can be positively correlated with lipid solubility or negatively correlated with hydrogen bonding (29). The extent to which a compound forms hydrogen bonds is vital for its ability to permeate endothelial cell membranes. The higher the hydrogen bonding potential, lower the uptake into the brain. By reducing the hydrogen bonding potential for a congeneric series of steroid hormones, there was a log increase in uptake with each removal of hydrogen bond pairs. The correlation of blood-brain distribution coefficients (as log BB in-vivo and in-vitro values) using hydrogen bonding descriptors are available (30) but are not very similar to correlations for log PS. Hence the factors that influence blood-brain distribution are not quantitatively the same as those that influence brain perfusion. So it is vitally important when discussing brain uptake to specify what measure of brain uptake is being used. A variety of in silico models (31) and in vitro permeability assays (32) have been developed in an attempt to characterize and predict BBB permeability and integrate such prediction in the early phases of drug development, together with various other considerations (33-35).
In-vivo and In-vitro models to study drug transport across the blood-brain and blood-CSF barriers
The pharmacokinetics and pharmacodynamics of drugs in the CNS are understood by their unbound concentrations in the extracellular fluid of the brain. Various in-vivo and in-vitro techniques are available to study this property. The in-vivo techniques include the brain uptake index (BUI) (26), the brain efflux index (BEI) (36), brain perfusion (37), the unit impulse response method (38) and micro-dialysis (39).
The efflux transport across the BBB is a very important process for explaining the mechanism of the apparent restricted cerebral distribution of drugs after their systemic administration. In order to examine the BBB efflux transport mechanism under in-vivo conditions, the intracerebral microinjection technique has been developed and recently established as the BEI. The BEI value is defined as the relative percentage of drug effluxed from the ipsilateral (that is, they do not cross to the opposite hemisphere) cerebrum to the circulating blood across the BBB compared with the amount of drug injected into the cerebrum, i.e.
The advantages of the BEI method are its ability to allow determination of the apparent in vivo drug efflux rate constant across the BBB, monitoring the concentration dependency of the test drug and the performance of inhibition studies. By contrast, the limitations of the BEI method are that only one data point can be obtained for a single intracerebral microinjection. The drug concentration in the cerebrum cannot be accurately determined. In other words, at the present time, the drug concentration in the brain is estimated by using the dilution factor, i.e. 30.3- to 46.2-fold dilution (36).
The brain interstitial fluid (ISF) concentration is a determinant for the effect of a drug in the CNS in-vivo. If the drug would cross the BBB in significant quantities by passive diffusion, the brain ISF concentration will equal the plasma unbound drug concentration after its administration. In this case, the plasma unbound drug concentration will be very important in predicting the CNS effect. However, if the brain ISF concentration of a drug is significantly lower than the plasma unbound drug concentration, it will be very important to identify the mechanism involved. For the direct measurement of brain ISF drug concentration, many researchers have found brain micro-dialysis to be a useful technique (40, 41). Micro-dialysis is a method of choice in the study of in-vivo drug transport across the BBB, based on brain's physiological and anatomical characteristics considering it to be a non-homogeneous compartment. In addition, drug disposition in the brain is determined by protein binding, blood flow, BBB transport, and the exchange between brain extracellular fluid (ECF) and brain cells. Nevertheless, intra-cerebral micro-dialysis is an invasive technique: it involves the implantation of a probe, which may cause tissue trauma, and hence may have consequences for BBB function. Therefore it is necessary to determine whether intra-cerebral micro-dialysis provides meaningful data on drug transport across the BBB and drug disposition in the brain.
Since thousands of new therapeutic compounds will have to be tested in the near future; alternatives to in-vivo test systems must be developed. Thus, in-vitro models that closely mimic the in-vivo system, at least with respect to barrier properties, are in high demand. Blood-brain barrier models now available make use of cerebral capillary endothelium (porcine brain capillary endothelial cells) or choroid plexus epithelial cells (porcine choroid plexus) (42, 43). Both cell types need serum in the growth medium to proliferate. Serum, however, inhibits the formation of tight cell-cell contacts. Withdrawal of serum favors cellular polarity and increases the barrier properties drastically. Electrical resistance is an easy measure of junctional tightness (44). A very sophisticated but highly reliable and reproducible new method is impedance spectroscopy (IS) (45), in which AC potentials are applied over a wide frequency range. At a single fixed frequency, AC potentials may be applied and analyzed if only relative changes after substrate application are expected. IS yields information about both conductivity and dielectric constant (capacitance) of the interfacial region of the cell monolayer. Essentially three types of brain capillary endothelial cell culture are currently used by researchers: primary cultures, cell lines and co-culture systems. The limitation of primary cultures has been their higher para-cellular permeability, reflected by the measurement of the electrical resistance across the monolayer. Later developments led to the generation of rat, bovine and human immortalized endothelial cells and their use as a replacement for primary cells in in-vitro BBB models (46). However, these cell systems have not been characterized to the same extent as either primary or passaged cells. The in-vitro BBB model, consisting of a co-culture of brain capillary endothelial cells on one side of a filter and astrocytes on the other, is currently used. The strong correlation between the in-vivo and in-vitro values demonstrated that this in-vitro system is an important tool for the investigation of the role of the BBB in the delivery of nutrients and drugs to the CNS (47). The main advantage of this model is the possible rapid evaluation of strategies for achieving drug targeting to the CNS or to appreciate the eventual central toxicity of systemic drug and to elucidate the molecular transport mechanism of substances across the BBB.
STRATEGIES FOR ENHANCED CNS DRUG DELIVERY
To circumvent the multitude of barriers inhibiting CNS penetration by potential therapeutic agents, numerous drug delivery strategies have been developed (6, 9, 15, 48-50). These strategies generally fall into one or more of the following three categories: manipulating drugs, disrupting the BBB and finding alternative routes for drug delivery.
Drug Manipulations
Lipophilic Analogs
CNS penetration is favored by low molecular weight, lack of ionization at physiological pH, and lipophilicity (13). Delivery of poorly lipid-soluble compounds to the brain requires some way of getting past the BBB. There are several possible strategies, such as transient osmotic opening of the BBB, exploiting natural chemical transporters, high-dose chemotherapy, or even biodegradable implants. But all of these methods have major limitations: they are invasive procedures, have toxic side effects and low efficiency, and are not sufficiently safe. Heroin, a diacyl derivative of morphine, is a notorious example that crosses the BBB about 100 times more easily than its parent drug just by being more lipophilic. Hence, a possible strategy is to smuggle compounds across as their lipophilic precursors. Because drug's lipophilicity correlates so strongly with cerebro-vascular permeability, hydrophobic analogues of small hydrophilic drugs ought to more readily penetrate the BBB. This strategy has been frequently employed, but the results have often been disappointing. The best examples of such attempts are the series of lipophilic analogues of nitrosoureas where a quantitative structural activity relationship (QSAR) study indicated the anti-neoplastic activity was inversely proportional to their lipophilicity. This is because the more lipophilic analogs becomes less soluble in the aqueous plasma and bind more readily to plasma proteins, leading to lower concentrations of drug available for diffusion into the CNS and demonstrate diminished alkylating activity and increased dose limiting toxicity. Hence, when a drug is delivered via the circulatory system for the treatment of CNS diseases, a delicate balance between cerebro-vascular permeability and plasma solubility is required. Specifically, the optimal log Po/w is approximately 1.5 to 2.5 (51). However, log Po/w alone seems to have a more limited performance in predicting brain/blood concentration ratios, but in combination with other parameters can still reasonably predict brain-blood partitioning (52, 53).
A second strategy for increasing the lipophilicity of a hydrophilic therapeutic agent is to surround the hydrophilic molecule with a sphere of lipids in the form of a liposome. The strategies for linking drugs to transport vectors shown in Table 2 involve an approximate 1:1 stoichiometry of vector to drug.
Table 2: Diversity in strategies for linking drugs to transport vectors.
However, the carrying capacity of the vector could be greatly expanded by incorporation of the non-transportable drug in liposomes, followed by subsequent conjugation of the liposome to a BBB drug delivery vector. Liposomes, even small unilamellar vesicles, do not undergo significant transport through the BBB in the absence of vector-mediated drug delivery (54). Another problem with liposomes is that these structures are rapidly removed from the bloodstream following intravenous administration, owing to uptake by cells lining the reticulo-endothelial system. The dual problems of mediating BBB transport and inhibiting peripheral clearance of liposomes were solved by the combined use of PEGylation technology and chimeric peptide technology (54). In this construct, a novel bi-functional PEG 2000 derivative that contains a maleimide at one end (for attachment to a thiolated MAb [murine monoclonal antibody]) and a distearoylphosphatidylethanolamine (DSPE) moiety at the other end (for incorporation into the liposome surface) was used to prepare the PEGylated immunoliposomes. The combined use of PEGylation technology, liposome technology, and chimeric peptide technology results in the construction of PEGylated immuno-liposomes that are capable of receptor-mediated transport through the BBB in-vivo (55). MAb binds to the BBB transferrin receptor, and it has been successfully used as a vector in delivery of other large molecules across the BBB. Since, a single liposome may carry up to 10,000 drug molecules, the immuno-liposome delivery system has the ability to dramatically increase brain drug delivery by up to four orders of magnitude. This delivery system may be of significance to brain drug delivery because it permits brain targeting of the liposomally encapsulated drug, and may consequently offer a significant reduction in side effects. Compounds with excellent neuro-pharmacologic potential in-vitro, which may have been rejected for clinical use because of low brain delivery (or some minor side-effects) may now be reevaluated for potential use in conjunction with this delivery system. Since the liposome capsule undergoes degradation to release its contents, the drug is delivered without the use of disulfide or ester linkages, which may significantly affect pharmacological actions (54). This micro-encapsulation strategy, and the use of living cells developed to produce neuro-pharmacological agents (56), is regarded as two of the more promising recent developments in brain drug delivery (57).
Prodrugs
Brain uptake of drugs can be improved via prodrug formation (58). Prodrugs are pharmacologically inactive compounds that result from transient chemical modifications of biologically active species. The chemical change is usually designed to improve some deficient physicochemical property, such as membrane permeability or water solubility. After administration, the prodrug, by virtue of its improved characteristics, is brought closer to the receptor site and is maintained there for longer periods of time. Here it gets converted to the active form, usually via a single activating step. For example, esterification or amidation of hydroxy-, amino-, or carboxylic acid- containing drugs, may greatly enhance lipid solubility and, hence, entry into the brain. Once in the CNS, hydrolysis of the modifying group will release the active compound. Unfortunately, simple prodrugs suffer from several important limitations. Going to extremes on the lipophilic precursor scale, a possible choice for CNS prodrugs is coupling the drug to a lipid moiety, such as fatty acid, glyceride or phospholipids. Such prodrug approaches were explored for a variety of acid-containing drugs, like levodopa, GABA, Niflumic acid, valproate or vigabatrin are coupled to diglycerides or modified diglycerides (59). While increased lipophilicity may improve movement across the BBB, it also tends to increase uptake into other tissues, causing an increased tissue burden. This selectivity in delivery is especially detrimental when potent drugs such as steroids or cytotoxic agents are considered, since toxicity is exacerbated at non-target sites. Moreover, while increased lipophilicity may facilitate drug uptake into the CNS, it also enhances efflux processes. This can result in poor tissue retention and short biological action. Furthermore, while the only metabolism associated with a prodrug should be its conversion to the parent drug, other routes can occur, and the formed metabolites may contribute to the toxicity of the compounds. These effects, that is poor selectivity, poor retention, and the possibility for reactive metabolites, may often conspire to decrease, not to increase, the therapeutic index of drugs masked as prodrugs. On the other hand, prodrug approaches that target specific membrane transporters have also been explored more recently (chemically) transforming the drug to be delivered so that it can become the subject of some specific membrane transporter, such as the amino acids, peptide or glucose transporters (60).
Chemical Drug Delivery
Chemical drug delivery systems (CDDS) represent novel and systematic ways of targeting active biological molecules to specific target sites or organs based on predictable enzymatic activation. They are inactive chemical derivatives of a drug obtained by one or more chemical modifications so that the newly attached moieties are monomolecular units (generally comparable in size to the original molecule) and provide a site-specific or site-enhanced delivery of the drug through multi-step enzymatic and/or chemical transformations. During the chemical manipulations, two types of bio-removable moieties are introduced to convert the drug into an inactive precursor form. A targetor (T) moiety is responsible for targeting, site-specificity, and lock-in, while modifier functions (F1... Fn ) serve as lipophilizers, protect certain functions, or fine-tune the necessary molecular properties to prevent premature, unwanted metabolic conversions. The CDDS is designed to undergo sequential metabolic conversions, disengaging the modifier functions and finally the targetor, after this moiety fulfils its site- or organ-targeting role. Undoubtedly, the concept evolved from the prodrug concept, but became essentially different by the introduction of multi-step activation and targetor moieties. Within the present formalism, one can say that prodrugs contain one or more F moieties for protected or enhanced overall delivery, but they do not contain T moieties. Brain-targeting chemical delivery systems represent just one class of CDDS; however, this is the most developed class. Using the general CDDS concept, successful deliveries have been achieved to the brain, to the eye, and to the lung (61).
These CDDS are based on the idea that, if a lipophilic compound that enters the brain is converted there into a lipid-insoluble molecule, it will no longer be able to come out, i.e. it will become `locked- in'. If the same conversion also takes place in the rest of the body, it accelerates peripheral elimination and improves targeting. In principle, many targetor moieties are possible for a general system of this kind, but the one based on the 1,4-dihydrotrigonelline´trigonelline (coffearine) system, where the lipophilic 1,4-dihydro form (T) is converted in-vivo to the hydrophilic quaternary form (T*), proved the most useful. This conversion takes place easily everywhere in the body since it is closely related to that of the ubiquitous NAD(P)H´NAD(P)+ coenzyme system associated with numerous oxidoreductases and cellular respiration. Since, oxidation takes place with direct hydride transfer and without generating highly active or reactive radical intermediates, it provides a nontoxic targetor system. Furthermore, since for small quarternary pyridinium ions rapid elimination from the brain, probably due to involvement of an active transport mechanism that eliminates small organic ions, has been shown (62), the T+ moiety formed during the final release of the active drug D from the charged T -D form will not accumulate within the brain. Meanwhile, the charged T -D form is locked behind the BBB into the brain, but is easily eliminated from the body due to the acquired positive charge, which enhances water solubility. After a relatively short time, the delivered drug D (as the inactive, locked-in T+ -D) is present essentially only in the brain, providing sustained and brain-specific release of the active drug. It has to be emphasized that the system not only achieves delivery to the brain, but it provides preferential delivery, which means brain targeting. Ultimately, this should allow smaller doses and reduce peripheral side effects.
Furthermore, since the `lock-in' mechanism works against the concentration gradient, it provides more prolonged effects. Consequently, CDDSs can be used not only to deliver compounds that otherwise have no access to the brain, but also to retain lipophilic compounds within the brain, as has indeed been achieved, for example, with a variety of steroid hormones. During the last decade, the system has been explored with a wide variety of drug classes. In a recent addition to the drug-targeting arsenal, targeted drug delivery to the brain via phosphonate derivatives was also explored, and so-called anionic chemical delivery systems (aCDDS) were designed, synthesized, and evaluated for testosterone and zidovudine (63). Here, an (acyloxy) alkyl phosphonate-type targetor moiety is used, and formation of an anionic 2 intermediate (T- -D) is expected to provide the `lock-in'. In addition, molecular packaging, an extension of the CDDS approach, achieved the first documented noninvasive brain delivery of neuropeptides in pharmacologically significant amounts. In this approach the peptide unit is part of a bulky molecule, dominated by lipophilic modifying groups that direct BBB penetration and prevent recognition by peptidases (64-67). Such a brain targeted packaged peptide delivery system contains the following major components: the redox targetor (T); a spacer function (S), consisting of strategically used amino acids to ensure timely removal of the charged targetor from the peptide; the peptide itself (P); and a bulky lipophilic moiety (L) attached through an ester bond or sometimes through a C- terminal adjuster (A) at the carboxyl terminal to enhance lipid solubility and to disguise the peptide nature of the molecule. To achieve delivery and sustained activity with such complex systems, it is very important that the designated enzymatic reactions take place in a specific sequence. Upon delivery, the first step must be the conversion of the targetor to allow for `lock-in'. This must be followed by removal of the L function to form a direct precursor of the peptide that is still attached to the charged targetor. Subsequent cleavage of the targetor-spacer moiety finally leads to the active peptide.
Another method called redox chemical delivery systems involves linking a drug to the lipophilic dihydropyridine carrier, creating a complex that after systemic administration readily transverses the BBB because of its lipophilicity. Once inside the brain parenchyma, the dihydropyridine moiety is enzymatically oxidized to the ionic pyridinium salt. The acquisition of charge has the dual effect of accelerating the rate of systemic elimination by the kidney and bile and trapping the drug-pyridinium salt complex inside the brain. Subsequent cleavage of the drug from the pyridinium carrier leads to sustained drug delivery in the brain parenchyma (68). This methodology increases intracranial concentrations of a variety of compounds, including neurotransmitters, antibiotics, and antineoplastic agents. This methodology has been extended to deliver neuroactive peptides such as enkephalin to the brain and has demonstrated promise in laboratory models, and evaluation of clinical efficacy in neurological patients is awaited with interest (69). These approaches should be useful in medicinal chemistry and research on drug delivery to the brain.
Carrier Mediated Drug Delivery
Carrier-mediated transport (CMT) and receptor-mediated transport (RMT) pathways are available for certain circulating nutrients or peptides. The availability of these endogenous CMT or RMT pathways means that portals of entry to the brain for circulating drugs are potentially available. In the brain capillary endothelial cells, which make up the BBB, there are several transport systems for nutrients and endogenous compounds (70, 71). They are (a) the hexose transport system for glucose and mannose, (b) the neutral amino acid transport system for phenylalanine, leucine and other neutral amino acids, (c) the acidic amino acid transport system for glutamate and aspartate, (d) the basic amino acid transport system for arginine and lysine, (e) the b-amino acid transport system for b-alanine and taurine, (f) the monocarboxylic acid transport system for lactate and short-chain fatty acids such as acetate and propionate, (g) the choline transport system for choline and thiamine, (h) the amine transport system for mepyramine, (i) the nucleoside transport system for purine bases such as adenine and guanine, but not pyrimidine bases, and (j) the peptide transport system for small peptides such as enkephalins, thyrotropin-releasing hormone, arginine-vasopressin etc. (71, 72). Utilization of differences in the affinity and the maximal transport activity among these transport systems expressed at the BBB is an attractive strategy for controlling the delivery and retention of drugs into the brain. These protein macromolecular carrier systems are characterized by saturability and molecular selectivity. The large neutral amino acids (LNAA) carrier system in the cerebro-vascular membrane is capable of transporting numerous endogenous as well as exogenous LNAAs, with great structural diversity; this characteristic has made it as an attractive strategy for CNS drug delivery (1). Levodopa, an exogenous precursor of dopamine, has a high affinity for the LNAA carrier system after traversing the antiluminal membrane of the cerebral endothelium where levodopa is decarboxylated to yield dopamine, which does not cross the BBB to an appreciable extent (51). A newly synthesized analog of melphalin, an antineoplastic agent, D,L- NAM, demonstrates enhanced affinity for the LNAA carrier (73), resulting in enhanced penetration via the LNAA carrier system. The peptide transporters existing at the BBB and their utilization for the specific brain delivery of small peptides or peptide-mimetic drugs remains to be fully investigated.
Receptor/Vector Mediated Drug Delivery
Receptor-mediated drug delivery to the brain employs chimeric peptide technology, wherein a non-transportable drug is conjugated to a BBB transport vector. The latter is a modified protein or receptor-specific monoclonal antibody that undergoes receptor-mediated transcytosis through the BBB in-vivo. Conjugation of drug to transport vector is facilitated with chemical linkers, avidin-biotin technology, polyethylene glycol linkers, or liposomes. Multiple classes of therapeutics have been delivered to the brain with the chimeric peptide technology, including peptide-based pharmaceuticals, such as a vasoactive peptide analog or neurotrophins such as brain-derived neurotrophic factor, anti-sense therapeutics including peptide nucleic acids (PNAs), and small molecules incorporated within liposomes (74, 75). The attachment of the drug that normally does not undergo transport through the BBB to a BBB transport vector such as the MAb, results in the formation of a chimeric peptide, provided the bifunctionality of the conjugate is retained (76). That is, the chimeric peptide must have not only a BBB transport function, but also a pharmaceutical function derived from the attached drug. Certain drugs may not be pharmacologically active following attachment to a BBB transport vector. In this case, it may be desirable to attach the drug to the transport vector via a cleavable disulfide linkage that ensures the drug is still pharmacologically active following release from the transport vector owing to cleavage of the disulfide bond. Depending on the chemistry of the disulfide linker, a molecular adduct will remain attached to the drug following disulfide cleavage, and the molecular adduct must not interfere with drug binding to the drug receptor (77). A second consideration with respect to the use of a disulfide linker is that virtually all of the cell disulfide reducing activity may be contained within the cytosol (78). Therefore, the chimeric peptide must undergo endosomal release following receptor-mediated endocytosis into the target brain cell, in order to distribute to the reductase compartment.
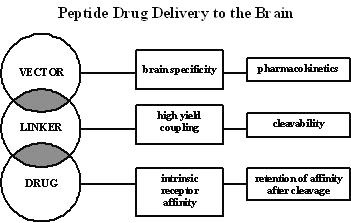
A second approach is to attach the drug to the transport vector via a non-cleavable linkage such as an amide bond. In this context, cleavability refers to reduction of the disulfide bond, since all the bonds including amide bonds are ultimately hydrolyzed in the lysosomal compartment. For certain peptide-based therapeutics if (a) a disulfide linker is not desired, and (b) the drug is not biologically active following conjugation via the amide linker, the PEGylation technology is used (Table 2) with a longer spacer arm comprised of a PEG moiety having a molecular mass of 2000-3400. With the PEG linker, the number of atoms comprising the linker is increased from 14 to _100. The placement of this long spacer arm between the transport vector and the drug releases any steric hindrance caused by attachment of the drug to the transport vector, and drug binding to the cognate receptor is not impaired (79). These considerations illustrate the multiplicity of approaches for linking drugs to transport vectors (Table 2 & Fig. 2), and the availability of these multiple approaches allows for designing transport linkers to suit the specific functional needs of the therapeutic under consideration.
Figure 2: Three interwoven areas of vector, linker and drug development with the corresponding criteria for optimization of each segment.
A summary of the different approaches for linking drugs to transport vectors is given in Table 2, and these approaches may be broadly classified as belonging to one of three classes: chemical, avidin-biotin, or genetic engineering. The chemical-based linkers employ activating reagents such as m-maleimidobenzoyl N-hydroxysuccinimide ester (MBS) or 2-iminothiolane (Traut's reagent), which activate primary amino groups on surface lysine (Lys) residues of either the drug or the transport vector (Table 2). This results in the formation of a stable thioether linkage which is comprised of only a single sulfur atom and is not subject to disulfide cleavability (79).
The concept of receptor-mediated transcytosis (RMT) of peptides through the BBB originated in the mid-1980s by means of the human BBB insulin receptor-mediated endocytosis of insulin into the brain capillary endothelium in-vitro and the transcytosis of insulin through the BBB in-vivo (80). Receptor-mediated transcytosis of insulin-like growth factors (IGFs) was demonstrated, and is consistent with the earlier observations that, like insulin, IGF-1 and IGF-2 are bound and endocytosed by animal and human brain capillaries in a receptor-mediated mechanism (80). Recently, a specific receptor for leptin has been characterized using human brain capillaries (81). Leptin is synthesized in peripheral tissues (fat) and is taken up by brain to induce satiety via receptor mediated transcytosis through the BBB.
Adsorptive-mediated transcytosis (AME), a mechanism of brain uptake that is related to receptor-mediated transcytosis, operates for peptides and proteins with a basic isoeletric point (cationic proteins) and for some lectins (glycoprotein-binding proteins). The initial binding to the luminal plasma membrane is mediated by electrostatic interactions with anionic sites or by specific interactions with sugar residues, respectively. In order to establish the structural specificity of AME at the BBB, uptake of several synthetic peptides having various molecular sizes, basicities and hydrophobicities, and carboxyl-terminal structures was compared by using primary cultured bovine endothelial cells. These results indicated that not the number of constituent amino acids of peptides, but rather the C-terminal structure and the basicity of the molecules, are important determinants of uptake by the AME system at the BBB (82).
Nanoparticles have also been used as transport vectors for peptides. Nanoparticles consist of colloidal polymer particles of poly-butylcyanoacrylate with the desired peptide absorbed onto the surface and then coated with polysorbate 80. Nanoparticles have been used as a vector for delivery of hexapeptide dalargin (an enkephalin analog). Intravenous injections of the vector-dalargin produce analgesia, while dalargin alone does not (83). Drugs that have successfully been transported across the BBB with the nanoparticles include loperamide, tubocerarine and doxorubicin (84, 85). The mechanism of nanoparticle transport has not yet been fully elucidated. The most probable transport pathway seems to be endocytosis by the blood capillary endothelial cells following adsorption of blood plasma components, most likely apolipoprotein E (apo E), after intravenous injection. These particles interact with the Low Density Lipoproteins (LDL) receptors on the endothelial cells and then get internalized. After internalization by the brain capillary endothelial cells, the drug releases in these cells by desorption or degradation of the nanoparticles and diffuses into the residual brain. Alternatively, transport may occur by transcytosis of the nanoparticles with drug across the endothelial cells (86). Per-coating of nanoparticles with polysorbate led to adsorption of apo E and possibly other plasma components, which seem to be able to interact with the LDL receptors on the brain endothelial cells, which could lead to their endocytosis (87). In addition to these processes, polysorbates seem to be able to inhibit the efflux pump. This inhibition could contribute to the brain transport properties of the nanoparticles (88). However the possibility of a general toxic effect is also a serious impediment (89).
Disturbing the Blood-Brain Barrier
Despite recent developments for enhanced CNS penetration, the BBB remains a formidable obstacle that compromises successful treatment of many neurological disorders. The second invasive strategy for enhanced CNS drug delivery involves the systemic administration of drugs in conjunction with transient BBB disruption (BBBD). Theoretically, with the BBB weakened, systemically administered drugs can undergo enhanced extravasation rates in the cerebral endothelium, leading to increased parenchymal drug concentrations. A variety of techniques that transiently disrupt the BBB have been investigated; however, albeit physiologically interesting, many are unacceptably toxic and therefore not clinically useful. These include the infusion of solvents such as dimethyl sulfoxide or ethanol and metals such as aluminium; X-irradiation; and the induction of pathological conditions including hypertension, hypercapnia, hypoxia or ischemia. The mechanisms responsible for BBBD with some of these techniques are not well understood. A somewhat safer technique involves the systemic delivery of the convulsant drug, metrazol, which transiently increases the BBB permeability while causing seizures. Concurrent administration of the anticonvulsant pentobarbital blocks seizing while allowing BBBD to persist. The BBB can also be compromised by the systemic administration of several antineoplastic agents including VP-16, cisplatin, hydroxylurea, flurouracil and etoposide.
Osmotic Blood-Brain Barrier Disruption
In the search for treatment of patients with rapidly growing, high grade gliomas, osmotic opening of the BBB was developed. Intracarotid injection of an inert hypertonic solution such as mannitol or arabinose has been employed to initiate endothelial cell shrinkage and opening of BBB tight junctions for a period of a few hours, and this permits delivery of antineoplastic agents to the brain (90). Though this treatment is still investigational, the fact that some patients who fail systemic chemotherapy have responded to similar or lower doses of intracarotid drugs is an often-cited argument in favor of the method. One reason for the unfavorable toxic/therapeutic ratio often observed with hyperosmotic BBBD is that this methodology results in only a 25% increase in the permeability of the tumor microvasculature, in contrast to a 10-fold increase in the permeability of normal brain endothelium. Although controversial, the method has shown promise in augmenting delivery of neurotoxic drugs to the CNS (91). However, some glial tumors have an endothelial barrier which is compromised, probably because the glial production of barrier-inducing factors is altered. For this reason, osmotic opening used in conjunction with cytotoxic drugs (such as carboplatin) may give an advantage over traditional chemotherapy. Osmotic disruption of the BBB has also been suggested as a delivery strategy for recombinant adenoviral vectors for gene transfer to intracerebral tumors (92), and for magnetic resonance imaging agents for diagnosis of brain metastases using iron oxide conjugates (93), but there are problems which must be overcome before the routine clinical use of this technique can be realized (94). Osmotic disruption seems to be most successful in treating primary non-AIDS CNS lymphoma (95). As a possible alternative to osmotic disruption of the BBB, Kaya et al. (96) have shown that 20-30% of the total brain microvessels become the more permeable fenestrated capillaries after induction through prolonged (4 week) infusions of either retinoic acid (100 mM) or phorbol myristate acetate (PMA) (150 ng/ ml). The chemical induction of fenestrated capillaries is attributed to the production of the plasminogen activator urokinase, and is completely reversed 1-2 months after delivery of retinoic acid or PMA is stopped (96). Osmatic distruption also has been tested as a strategy for the delivery of macromolecular drugs such as monocolonal antibodies, nanoparticles and viruses (97-99). However, the procedure breaks down the self-defense mechanism of the brain and levels it vulnerable to damage or infection from all circulating chemicals or toxins. The risk factors include, the passage of plasma proteins, the altered glucose uptake, the expression of heat shock proteins, microembolism or abnormal neuronal function (100).
Biochemical Blood-Brain Barrier Disruption
Recently, new and potentially safer biochemical techniques have been developed to disrupt the BBB. Selective opening of brain tumor capillaries (the blood-tumor barrier), by the intracarotid infusion of leukotriene C4 was achieved without concomitant alteration of the adjacent BBB (101). In contrast to osmotic disruption methods, biochemical opening utilizes the novel observation that normal brain capillaries appear to be unaffected when vasoactive leukotriene treatments are used to increase their permeability. However, brain tumor capillaries or injured brain capillaries appear to be sensitive to treatment with vasoactive leukotrienes, and the permeation is dependent on molecular size. The mechanism was shown to be related to the abundance of g-glutamyl transpeptidase (g-GTP) in normal brain capillaries; this enzyme requires glial inductive influence for its expression, and it is down- regulated in tumors, resulting in a reduction of the enzymatic barrier in tumor endothelial cells (102). From this origin, studies of the effects of alternative vasoactive amines were initiated, and it has been demonstrated that bradykinin, histamine and the synthetic bradykinin analog RMP-7 (receptor-mediated permeabilizer) infusion also selectively open the blood tumor barrier in experimental animals. The responsible biochemical mechanism has yet to be elucidated, but it has been established that the effect of the bradykinin analog RMP-7 is mediated specifically through bradykinin B2 receptors. Enhanced tumor drug delivery and survival in glioma bearing rats have also been seen with RMP-7 (103). These findings were so promising that clinical trials were initiated using the bradykinin analog RMP-7 to enhance brain delivery of antitumor medications. In the current Phase II multinational clinical trials, intravenous or intra-arterial RMP-7, is being administered together with carboplatin in the treatment of human gliomas, (104, 105) but now abandoned for the same reasons as the osmotic BBB disruption approach (100).
Alternative Routes to CNS Drug Delivery
Despite advances in rational CNS drug design and BBBD, many potentially efficacious drug molecules still cannot penetrate into the brain parenchyma at therapeutic concentrations. A third class of strategies aimed at enhancing CNS penetration of drug molecules is composed of delivery methodologies that do not rely on the cardiovascular system. These alternative routes for controlled CNS drug delivery obviate the need for drug manipulation to enhance BBB permeability and/or BBBD by circumventing the BBB altogether. Since, most aforementioned techniques aim to enhance the CNS penetration of drugs delivered via the circulatory system, the result is higher drug penetration throughout the entire body and frequently unwanted systemic side effects. Additionally, systemically administered agents must penetrate the BBB to enter the brain, which is a formidable task.
Intraventricular/Intrathecal Route
One strategy for bypassing the BBB that has been studied extensively both in laboratory and in clinical trials is the intralumbar injection or intreventricular infusion of drugs directly into the CSF. Drugs can be infused intraventricularly using an Ommaya reservoir, a plastic reservoir implanted subcutaneously in the scalp and connected to the ventricles within the brain via an outlet catheter. Drug solutions can be subcutaneously injected into the implanted reservoir and delivered to the ventricles by manual compression of the reservoir through the scalp.
When compared to vascular drug delivery, intra-CSF drug administration theoretically has several advantages. Intra-CSF administration bypasses the BCB and results in immediate high CSF drug concentrations. Since, the drug is somewhat contained within the CNS, a smaller dose can be used, potentially minimizing systemic toxicity. Furthermore, drugs in the CSF encounter minimized protein binding and decreased enzymatic activity relative to drugs in plasma, leading to longer drug half-life in the CSF. Finally, because the CSF freely exchanges molecules with the extracellular fluid of the brain parenchyma, delivering drugs into the CSF could theoretically result in therapeutic CNS drug concentrations.
However, this delivery method has not lived up to its theoretical potential for several reasons. These include a slow rate of drug distribution within the CSF and increase in intracranial pressure associated with fluid injection or infusion into small ventricular volumes. It results in to high clinical incidence of hemorrhage, CSF leaks, neurotoxicity and CNS infections. The success of this approach is limited by the CSF-brain barrier, composed of barriers to diffusion into the brain parenchyma. Because the extracellular fluid space of the brain is extremely tortuous, drug diffusion through the brain parenchyma is very slow and inversely proportional to the molecular weight of the drug (106). For macromolecules, such as proteins, brain parenchymal concentrations following intra-CSF administration are undetectable (107, 108). For these reasons, intra-CSF chemotherapy in the treatment of intraparenchymal CNS tumors has not proven to be effective. The greatest utility of this delivery methodology has been in cases where high drug concentrations in the CSF and/or the immediately adjacent parenchyma are desired, such as in the treatment of carcinomatous meningitis or for spinal anesthesia/analgesia (109).
Intrathecal and intracerebral drug administration differs fundamentally from systemic drug administration in terms of pharmacokinetic characteristics determining brain tissue concentration, where the available dose reaching the target organ is 100%. However, there are large gradients inside the tissue with very high local concentrations at the site of administration (the ventricular surface or tissue site of injection) and zero concentration at some distance for macromolecules. Since, they have low diffusion coefficients, the gradients will be even steeper than what has been measured for small molecular weight drugs (110, 111). After intracerebroventricular (icv) injection, the rate of elimination from the CNS compartment is dominated by cerebrospinal fluid dynamics. Clinical examples of intrathecal small drug delivery are the icv administration of glycopeptide and aminoglycoside antibiotics in meningitis, the intraventricular treatment of meningeal metastasis, intrathecal injection of baclofen for treatment of spasticity and the infusion of opioids for severe chronic pain. These examples have in common the fact that the drug targets in all instances are close to the ventricular surface. Superficial targets may also be accessible for some macromolecular drugs.
Olfactory Pathway
An alternative CNS drug delivery strategy that has received relatively little attention is the intranasal route. Drugs delivered intranasally are transported along olfactory sensory neurons to yield significant concentrations in the CSF and olfactory bulb. In recent studies, intranasal administration of wheat germ agglutinin horseradish peroxidase resulted in a mean olfactory bulb concentration in the nanomolar range. In theory, this strategy could be effective in the delivery of therapeutic proteins such as brain-delivered neurotropic factor (BDNF) to the olfactory bulb as a treatment for Alzheimer's disease (112). The nasal drug delivery to the CNS is thought to involve either an intraneuronal or extraneuronal pathway (49, 113). Recent evidence of direct nose-to-brain transport (114) and direct access to CSF of three neuropeptides bypassing the bloodstream has been shown in human trials, despite the inherent difficulties in delivery (113). The difficulties that have to be overcome include an enzymatically active, low pH nasal epithelium, the possibility of mucosal irritation or the possibility of large variability caused by nasal pathology, such as common cold. An obvious advantage of this method is that it is noninvasive relative to other strategies. In practice, however, further study is required to determine if therapeutic drug concentrations can be achieved following intranasal delivery.
Interstitial Delivery
The most direct way of circumventing the BBB is to deliver drugs directly to the brain interstitium. By directing agents uniquely to an intracranial target, interstitial drug delivery can theoretically yield high CNS drug concentrations with minimal systemic exposure and toxicity. Furthermore, with this strategy, intracranial drug concentrations can be sustained, which is crucial in treatment with many chemotherapeutic agents.
Injections, Catheters, and Pumps
Several techniques have been developed for delivering drugs directly to the brain interstitium. One such methodology is the Ommaya reservoir or implantable pump as discussed earlier under intraventricular/intrathecal route. This technique, however, does achieve truly continuous drug delivery. More recently, several implantable pumps have been developed that possess several advantages over the Ommaya reservoir. This can be implanted subcutaneously and refilled by subcutaneous injection and are capable of delivering drugs as a constant infusion over an extended period of time. Furthermore, the rate of drug delivery can be varied using external handheld computer control units. Currently each of the three different pumps available for interstitial CNS drug delivery operates by a distinct mechanism. The Infusaid pump uses the vapour pressure of compressed Freon to deliver a drug solution at a constant rate; the MiniMed PIMS system uses a solenoid pumping mechanism, and the Medtronic SynchroMed system delivers drugs via a peristaltic mechanism. The distribution of small and large drug molecules in the brain can be enhanced by maintaining a pressure gradient during interstitial drug infusion to generate bulk fluid convection through the brain interstitium (115) or by increasing the diffusion gradient by maximizing the concentration of the infused agent (116) as a supplement to simple diffusion. Another recent study shows that the epidural (EPI) delivery of morphine encapsulated in multivesicular liposomes (DepoFoam drug delivery system) produced a sustained clearance of morphine and a prolonged analgesia, and the results suggest that this delivery system is without significant pathological effects at the dose of 10mg/ml morphine after repeated epidural delivery in dogs (117).
Biodegradable polymer Wafers, Microspheres and Nanoparticles
Though interstitial drug delivery to the CNS has had only modest clinical impact, its therapeutic potential may soon be realized using new advances in polymer technologies to modify the aforementioned techniques. Polymeric or lipid-based devices that can deliver drug molecules at defined rates for specific periods of time are now making a tremendous impact in clinical medicine (118, 119). Drug delivery directly to the brain interstitium using polyanhydride wafers can circumvent the BBB and release unprecedented levels of drug directly to an intracranial target in a sustained fashion for extended periods of time. The fate of a drug delivered to the brain interstitium from the biodegradable polymer wafer was predicted by a mathematical model based on (a) rates of drug transport via diffusion and fluid convection; (b) rates of elimination from the brain via degradation, metabolism and permeation through capillary networks; and (c) rates of local binding and internalization (120). Such models are used to predict the intracranial drug concentrations that result from BCNU-loaded pCPP:SA (1,3 bis-para-carboxyphenoxypropane:sebacic acid) wafers as well as other drug-polymer combinations, paving the way for the rational design of drugs specifically for intracranial polymeric delivery.
Conjugation of a polymerically delivered chemotherapeutic agent to a water-soluble macromolecule increases drug penetration into the brain by increasing the period of drug retention in brain tissue (121). Hanes et al have recently developed IL-2-loaded biodegradable polymer microspheres for local cytokine delivery to improve the immunotherapeutic approach to brain tumor treatment (122). In theory, polymeric cytokine delivery has several advantages over delivery from transducted cells, including obviating the need for transfecting cytokine genes, producing longer periods of cytokine release in-vivo and yielding more reproducible cytokine release profiles and total cytokine dose. Microparticles can also be easily implanted by stereotaxy in discrete, precise and functional areas of the brain without damaging the surrounding tissue. This type of implantation avoids the inconvenient insertion of large implants by open surgery and can be repeated if necessary (123). The feasibility of polymer-mediated drug delivery by the standard chemotherapeutic agent 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) showed that local treatment of gliomas by this method is effective in animal models of intracranial tumors. This led to clinical trials for glioma patients, and subsequent approval of GliadelTM [(3.8% BCNU): p(CPP:SA)] by the FDA and other worldwide regulatory agencies. Obviously, such an invasive approach can only be useful in a very limited number of patients, but this approach has been shown to prolong survival in patients with recurrent glioblastoma multiform brain tumors (119). Nevertheless, because of diffusion problems, even in this case, the therapeutic agent is likely to reach only nearby sites (108).
Polymeric nanoparticles have been proposed as interesting colloidal systems that allow the enhancement of therapeutic efficacy and reduction of toxicity of large variety of drugs (124). Nanoparticles were found to be helpful for the treatment of the disseminated and very aggressive brain tumors. Intravenously injected doxorubicin-loaded polysorbate 80-coated nanoparticles were able to lead to 40% cure in rats with intracranially transplanted glioblastomas (84). Another Study shows that PEGylated PHDCA (n-hexadecylcyanoacrylate) nanoparticles made by PEGyalated amphiphilic copolymer penetrate into the brain to a larger extent than all the other tested nanoparticle formulations, without inducing any modification of the BBB permeability (125). And the result defines two important requirements to take into account in the design of adequate brain delivery systems, long-circulating properties of the carrier and appropriate surface characteristics to permit interactions with endothelial cells. Valproic acid-loaded nanoparticles showed reduced toxic side effects of valporate therapy, not by reducing the therapeutically necessary dosage but by inhibition of formation of toxic metabolites (126). In conclusion, the capacity of the biodegradable polymer delivery methodology to deliver drugs directly to the brain interstitium is vast.
Drug Delivery from Biological Tissues
Another strategy to achieve interstitial drug delivery involves releasing drugs from biological tissues. The simplest approach to this technique is to implant into the brain a tissue that naturally secretes a desired therapeutic agent. This approach has been most extensively applied to the treatment of Parkinson's disease (51). Transplanted tissue often did not survive owing to a lack of neovascular innervation. Recently the enhanced vascularization and microvascular permeability in cell-suspension embryonic neural grafts relative to solid grafts has been demonstrated (127). An alternative extension of this method is to use gene therapy to develop optimized biological tissue for interstitial drug delivery. Prior to implantation, cells can be genetically modified to synthesize and release specific therapeutic agents. The therapeutic potential of this technique in the treatment of brain tumor was demonstrated (128). The use of nonneuronal cells for therapeutic protein delivery to the CNS has recently been reviewed (129). The survival of foreign tissue grafts may be improved by advancements in techniques for culturing distinct cell types. Co-grafted cells engineered to release neurotropic factors with cells engineered to release therapeutic proteins may enhance the survival and development of foreign tissue (130).
Ideally it would be possible to perform in-vivo genetic engineering to cause specific endogenous brain tissue to express a desired protein, circumventing the ischemic and immunogenic complications encountered with the implantation of foreign tissue grafts. One such technique that has been successfully used for the treatment of CNS malignancies involves in-vivo tumor transduction with the herpes simplex thymidine kinase (HS-tk) gene followed by treatment with anti-herpes drug ganciclovir was achieved by intra-tumoral injection of retroviral vector-producing cells containing the HS-tk gene, rendering the transfected tumor cells susceptible to treatment with ganciclovir (131). Other vector systems used in CNS gene transfer studies include retroviruses, adenoviruses, adeno-associated viruses, encapluation of plasmid DNA into cationic liposomes and neutral and oligodendrial stem cells. Although this approach holds remarkable therapeutic potential in the treatment of CNS diseases, its efficacy has thus far been hindered by a number of obstacles: restricted delivery of vector systems across the BBB, inefficient transfection of host cells, nonselective expression of the transgene and deleterious regulation of the transgene by the host (132).
Conclusions
The treatment of CNS diseases is particularly challenging because the delivery of drug molecules to the brain is often precluded by a variety of physiological, metabolic and biochemical obstacles that collectively comprise the BBB, BCB and BTB. The present outlook for patients suffering from many types of CNS diseases remains poor, but recent developments in drug delivery techniques provide reasonable hope that the formidable barriers shielding the CNS may ultimately be overcome. Drug delivery directly to the brain interstitium has recently been markedly enhanced through the rational design of polymer-based drug delivery systems. Substantial progress will only come about, however, if continued vigorous research efforts to develop more therapeutic and less toxic drug molecules are paralleled by the aggressive pursuit of more effective mechanisms for delivering those drugs to their CNS targets.
Promising strategies/devices
One can aim for either modification of existing drugs to increase BBB penetration by promising strategies or develop a new chemical entity that already possess the desired permeability properties. Table - 3 summarizes the various technical approaches for drug delivery to CNS with its advantages and limitations.
Table 3: Drug delivery to CNS: Technical approaches, advantages and limitations.
The promising strategies that can be exploited to promote drug delivery to the CNS are:
- Liposomes targeting to the brain by exploiting receptor mediated transcytosis system (55),
- Nanoparticles for drug delivery across BBB (84, 85, 133, 134),
- Implantation within the brain of either genetically engineered cells secreting a drug or a polymeric matrix or reservoir containing the drug (118-120),
- Chemical delivery systems based on predictable enzymatic activation (63-69),
- Chimeric peptide technology, wherein a non-transportable drug is conjugated to a BBB transport vector (81, 82),
- Neuroproteomics approaches and gene therapy for CNS disorders (135).
Combinations of drug delivery strategies and techniques will also no doubt prove to be useful.
References
Pardridge, W.M., Peptide drug delivery to the brain. Raven Press, New York, U.S.A., 1991.
Begley, D.J., The bloodbrain barrier: principles for targeting peptides and drugs to the central nervous system. J Pharm Pharmacol, 48:136146, 1996.
Schlossauer, B. and Steuer, H., Comparative anatomy, physiology and in vitro models of the blood-brain and blood-retina barrier. Curr Med Chem, 2:175-186, 2002.
Crone, C., The bloodbrain barrier: a modified tight epithelium, in Suckling AJ: Rumsby MG: Bradbury MWB (eds), The BloodBrain Barrier in Health and Disease. Ellis Harwood, Chichester, pp 1740, 1986.
Brightman M., Ultrastructure of brain endothelium, in Bradbury MWB (ed) Physiology and pharmacology of the blood-brain barrier. Handbook of experimental pharmacology 103, Springer-Verlag, Berlin, pp 122, 1992.
Lo, E.H., Singhal, A.B., Torchilin, V.P., and Abbott N.J., Drug delivery to damaged brain, Brain Res Rev, 38:140-148, 2001.
Davson, H.; Segal, M.B., Physiology of the CSF and blood brain barriers. CRC Press, Florida, USA, 1995.
Brownless, J. and Williams, C.H., Peptidases, peptides and the mammalian blood-brain barrier, J Neuroche, 60:1089-1096, 1993
Witt KA, Gillespie TJ, Huber JD, Egleton, R.D., and Davis, T.P., Peptide drug modifications to enhance bioavailability and blood-brain barrier permeability, Peptides, 22:2329-2343, 2001.
Nabeshima, S., Reese, T.S., Landis, D.M. and Brightman, M.W., Junctions in the meninges and marginal glia. J Comp Neurol, 164:2 127-169, 1975.
Brightman, M.W., The intracerebral movement of proteins injected into blood and cerebrospinal fluid of mice, Prog Brain Res, 29:19-40, 1968.
Saito, Y. and Wright, E.M., Bicarbonate transport across the frog choroid plexus and its control by cyclic nucleotides, J Physiol, 336:635-648, 1983.
Pardridge, W.M., Recent advances in blood brain-barrier transport. Annu Rev Pharmacol Toxicol, 28:25-39, 1988.
Cornford, E.M., Braun, L.D., Oldendorf, W.H. and Hill, M.A., Comparison of lipid-mediated bloodbrain barrier penetrability in neonates and adults. Am J Physiol, 243:C161C168, 1982.
Siegal, T. and zylber-Katz, E., Strategies for increasing drug delivery to the brain: focus on brain lymphoma, Clin Pharmacokinet, 41:171-186, 2002.
Cole, S.P.C., Bhardwaj, G., Gerlach, J.H., McKemzie, J.G., Grant, C.E., Almquist, K.C., Stewart, A.J., Kurz, E.U., Duncan, A.M.V. and Deeley, R.G., Over expression of a transporter gene in a mulitdrug-resistant human lung cancer cell line. Science, 258:1650-1654, 1992.
Taylor, E.M., The impact of efflux transportersin the brain on the development of drugs for CNS disorders, Clin Pharmacokinet, 41:81-92, 2002.
Sadeque, A.J., Wandel, C., He, H., Shah, S., and Wood. A.J., Increased drug delivery to the brain by P-glycoprotein inhibition, Clin Pharmacol Ther, 68:231-237, 2000.
Salvolainen, J., Edwards, J.E., Morgan, M.E., McNamara, P.J., and Anderson, B.D., Effects of a P-glycoprotein inhibitor on brain and plasma concentrations of anti-human immunodeficiency virus drugs administered in combination in rats, Drug Metab Dispos, 30:479-482, 2002.
Fellner, S., Bauer, B., Miller, D.S., Schaffrik, M., Fankhanel, M., Spruss, T., Bernhardt, G., Graeff, C., Farber, L., Gschaidmeier, H., Buschauer, A., and Fricker, G., Transport of paclitaxel (Taxol) across the blood-brain barrier in vitro and in vivo, J Clin Invest, 110:1309-1318, 2002.
Gupta, S.P., QSAR studies on drugs acting at the central nervous system. Chem Rev, 89:1765-1800, 1989.
Hansch, C., Leo, A. and Hoekman, D., Exploring QSAR. Hydrophobic, Electronic, and Steric Constants. American Chemical Society, Washington, DC, 1995.
van de Waterbeemd, H., Smith, D.A., Beaumont, K. and Walker, DK., Property-based design: optimization of drug absorption and pharmacokinetics. J Med Chem, 44:1313-1333, 2001.
Lin, J.H. and Lu, A.Y., Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol Rev, 49:403-449, 1997.
Lewis, D.F.V. and Dickins, M., Substrate SARs in human P450s. Drug DIscov Today, 7:918-925, 2002.
Oldendorf, W.H., Measurement of brain uptake of radiolabeled substances using a tritiated water internal standard. Brain Res, 24:16291639, 1970.
Pardridge, W.M., Triguero, D., Yang, J. and Cancilla, P.A., Comparison of in-vitro and in-vivo models of drug transcytosis through blood-brain barrier. J Pharm Exp Ther, 253:884-891, 1990.
Levin, V.A., Relationship of octanol/water partition coefficient and molecular weight to rat brain capillary permeability. J Med Chem, 23:682684, 1980.
Cornford, E.M. and Oldendorf, W.H., Epilepsy and the blood brain barrier. Adv Neurol 44:787812, 1986.
Abraham, M.H., Chadha, H.S. and Mitchell, R.C., Hydrogen bonding. 33. Factors that influence the distribution of solutes between blood and brain. J Pharm Sci, 83:1257-1268, 1994.
Sippl, W., Computational approaches for the prediction of blood-brain barrier permeation, Curr Med Chem, 2:212-227, 2002.
de Boer, A.G. and Gaillard, P.J., In vitro models of blood-brain barrie: when to use which, Curr Med Chem, 2:203-209, 2002.
Mertsch, K. and Maas, J., Blood-brain barrier penetration and drug development from an industrial point of view, Curr Med Chem, 2:189-209, 2002.
Buchwald, P. and Bodor, N., Computer-aided drug design: the role of quantitative structure-property, structure-activity and structure-metabolism relationships (QSPR, QSAR, QSMR), Drugs Future, 27:577-588, 2002.
Kerns, E.H., Hightroughput physicochemical profiling for drug discovery. J Pharm Sci, 90:1838-1858, 2001.
Kakee, A., Terasaki, T. and Sugiyama, Y., Brain efflux index as a novel method of solute analyzing efflux transport at the blood-brain barrier. J Pharmacol Exp Ther, 277:1550-1559, 1996.
Begley, D.J., Squires, L.K., Zlokovic, B.V., Mitrovic, D.M., Hughes, C.C.W., Revest, P.A. and Greenwood, J., Permeability of the blood-brain barrier to the immunosuppressive cyclic peptide cyclosporine A. J Neurochem ,55:1222-1230, 1998.
Jaehde, U., Langemeijer, M.W.E., de Boer, A.G. and Breimer, D.D., Cerebrospinal fluid transport and deposition of the quinolones ciprofloxacin and pefloxacin. J Pharmacol Exp Ther, 263(3):1140-1146, 1992.
de lange, E.C.M., de Boer, A.G. and Breimer, D.D., Monitoring in-vivo BBB drug transport: CSF sampling, the unit impulse response method and, with special reference, intracerebral microdialysis. STP Pharm Sci, 7(1):17-28, 1997.
Terasaki, T., Deguchi, Y., Kasama, Y., Pardridge, W.M. and Tsuji, A., Determination of in vivo steady-state unbound drug concentration in the brain interstitial fluid by microdialysis, Int J Pharm, 81:143152, 1992.
Menacherry, S., Hubert, W. and Justice, J.B., In vivo calibration of microdialysis probes for exogenous compounds, Anal Chem, 64:577583, 1992.
Tewes, B., Franke, H., Hellwig, S., Hoheisel, D., Decker, S., Griesche, D., Tilling, T., Wegener, J. and Galla, H.J., Preparation of endothelial cells in primary cultures obtained from the brains of 6-month old pigs, in de Boer AG: Sutanto W (eds), Tramsport across the blood-brain barrier: In-vitro and in-vivo techniques. Academic publishers, Amsterdam, Harwood, pp 91-97, 1997.
Gath, U., Hakvoort, A., Wegener, J., Decker, S. and Galla, H.J., Porcine choroid
splexus cells in culture: Expression of polarized phenotype, maintenance of barrier properties and apical secretion of CSF-components. Eur J Cell Biol, 74:68-78, 1997.Erben, M., Decker, S., Franke, H. and Galla, H.J., Electrical resistance measurements on cerebral capillary endothelial cells: A new technique to study small surfaces. J Biochem Biophys Methods, 30:227-238, 1995.
Wegener, J., Sieber, M. and Galla, H.L., Impedance analysis of epithelial and endothelial cell monolayers cultured on gold surfaces. J Biochem Biophys Methods, 32:151-170, 1996.
Hurst, R.D. and Fritz, I.B., Properties of an immortalized vascular endothelial/glioma cell coculture model of the blood-brain barrier. J Cell Physiol, 167:81-88, 1996.
Dehauck, M.P., Dehouck, B., Schluep, C., Lemaire, M. and Cecchelli, R., Drug transport to the brain: comparison between in-vitro and in-vivo models of the blood-brain barrier. Eur J Pharm Sci, 3:357-365, 1995.
Habgood, M.D., Begley, D.J. and Abbott, N.J., Determinants of passive drug entry into the central nervous system, Cell Mol Neurobiol, 20:231-253, 2000.
Thorne, R.G. and Frey II, W.H., Delivery of neurotropic factore to the central nervous system: pharmacokinetic consideration, Clin Pharmacokinet, 40:907-946, 2001.
Filmore, D., Breeching the blood-drain barrier, Modern Drug Discov, 5:22-27, 2002.
Madrid, Y., Langer, L.F., Brem, H. and Langer, R., New directions in the delivery of drugs and other substances to the central nervous system. Adv Pharmacol, 22:299-324, 1991.
Crivori, P., Cruciani, G. and CarruptP., Predicting blood-brain barrier permeation from three-dimensional molecular structure, J Med Chem, 39:4750-4755, 2000.
Iyer, M., Mishra, R. and Han, Y., Predicting blood-brain barrier partitioning of organic molecules using membrane-interaction QSAR analysis, Pharm Res, 19:1611-1621, 2002.
Huwyler, J., Wu, D. and Pardridge, W.M., Brain drug delivery of small molecules using immunoliposomes. Proc Natl Acad Sci, USA, 93:1416414169, 1996.
Huwyler, J., Yang, J. and Pardridge, W.M., Targeted delivery of daunomycin using immunoliposomes: pharmacokinetics and tissue distribution in the rat. J Pharmacol Exp Ther, 282:15411546, 1997.
Deglon, N., Heyd, B., Tan, S.A., Joseph, J.M., Zurn, A.D. and Aebischer, P., Central nervous system delivery of recombinant ciliary neurotrophic factor by polymer encapsulated differentiated C2C12 myoblasts. Hum Gene Ther, 7:21352146, 1996.
Maysinger, D. and Morinville, A., Drug delivery to the nervous system. Trends Biotechnol, 15:410418, 1997.
Bodor, N. and Kaminski, J.J., Prodrugs and site-specific chemical delivery systems. Annu Rep Med Chem, 22:303313, 1987.
Lambert, D.M., Rationale and applications of lipids as prodrug carriers. EurJ Pharm Sci, 11:S15-27, 2000.
Han, H.k and Amidon, G.L., Targeted prodrug design to optimize drug delivery, AAPS PharmSci, 2:E6, 2000.
Bodor, N. and Buchwald, P., Drug targeting via retrometabolic approaches. Pharmacol Ther 76:127, 1997.
Palomino, E., Kessel, D. and Horwitz, J.P., A dihydropyridine carrier system for sustained delivery of 1, 3-dideoxynucleosides to the brain. J Med Chem, 32:622625, 1989.
Somogyi, G., Nishitani, S., Nomi, D., Buchwald, P., Prokai, L. and Bodor, N., Targeted drug delivery to the brain via phosphonate derivatives. I: Design, synthesis, and evaluation of an anionic chemical delivery system for testosterone. Int J Pharm, 166:1526, 1998.
Boder, N., Prokai, L. and Wu, W-M., A strategy for delivering peptides into the central nervous system by sequential metabolism, Science, 257:1698-1700, 1992.
Bodor, N. and Prokai, L., Molecular packaging: peptide delivery to the central nervous system by sequential metabolism, in Taylor M: Amidon G (eds), Peptide-Based Drug Design: Controlling Transport and Metabolism. American Chemical Society, Washington, DC, pp 317337, 1995.
Chen, P., Bodor, N., Wu, W-M., and Prokai, L., Strategies to target kyotorphin analogues to the brain, J Med Chem, 41:3773-3781, 1998.
Wu, J., Yoon, S-H. Wu, W-M., and Bodor, N., Synthesis and biological evaluation of a brain targeted chemical delivery system of [Nva2]-TRH, J pharm Pharmacol, 54:945-950, 2002.
Bodor, N., Farag, H.H. and Brewster, M.E., Site-specific sustained release of drugs to the brain. Science, 214(18):13701372, 1981.
Bodor, N., Prokai, L., Wu, W.M., Farag, H.H., Jonnalagadda, S., Kawamura, M. and Simpkins, J., A strategy for delivering peptides into the central nervous system by sequential metabolism. Science, 257:16981700, 1992.
Pardridge, W.M., Transport of small molecules through the bloodbrain barrier: biology and methodology. Adv Drug Deliv Rev, 15:536, 1995.
Bergley, D.J., The bloodbrain barrier: principles for targeting peptides and drugs to the central nervous system. J Pharm Pharmacol, 48:136146, 1996.
Banks, W.A., Audus, K. and Davis, T.P., Permeability of the bloodbrain barrier to peptides: an approach to the development of therapeutically useful analogs. Peptides, 13:12891294, 1992.
Takada, Y., Vistica, D.T., Greig, N.H., Purdon, D., Rapoport, S.I. and Smith, Q.R., Rapid high-affinity transport of a chemotherapeutic amino acid across the bloodbrain barrier. Cancer Res, 52:21912196, 1992.
Pardridge, W.M., Vector-mediated drug delivery to the brain. Adv Drug Deliv Rev, 36:299321, 1999
Pardridge, W.M., Drug and gene targeting to the brain with molecular Trojan horses, Nat Rev Drug Discov, 1:131-139, 2002.
Pardridge, W.M., Peptide Drug Delivery to the Brain. Raven Press, NY, pp 1357, 1991.
Bickel, U., Yoshikawa, T., Landaw, E.M., Faull, K.F. and Pardridge, W.M., Pharmacologic effects in-vivo in brain by vector- mediated peptide drug delivery. Proc Natl Acad Sci, 90:26182622, 1993.
Lodish, H.F. and Kong, N., The secretory pathway is normal in dithiothreitol- treated cells, but disulfide-bonded proteins are reduced and reversibly retained in the endoplasmic reticulum. J Biol Chem, 268:2059820605, 1993.
Yoshikawa, T. and Pardridge, W.M., Biotin delivery to brain with a covalent conjugate of avidin and a monoclonal antibody the transferrin receptor. J Pharmacol Exp Ther, 263:897903, 1992.
Pardridge, W.M., Receptor-mediated peptide transport through the bloodbrain barrier. Endocrine Rev, 7:314330, 1986.
Golden, P.L., Maccagnan, T.J. and Pardridge, W.M., Human bloodbrain barrier leptin receptor: Binding and endocytosis in isolated human brain microvessels. J Clin Invest, 99:1418, 1997.
Tamai, I., Sai, Y., Kobayashi, H., Kamata, M., Wakamiya, T. and Tsuji, A., Structure-internalization relationship for adsorptive-mediated endocytosis of basic peptides at the bloodbrain barrier. J Pharmacol Exp Ther, 280:410415, 1997.
Kreuter, J., Alyautdin, R.N., Kharkevich, D.A. and Ivanov, A.A., Passage of peptides through the blood brain barrier with colloidal polymer particles (nanoparticles). Brain Res, 674:171174, 1995.
Kreuter, J., Nanoparticlate systems for brain delivery of drugs, Adv Drug Deliv Rev, 47:65-81, 2001.
Kreuter, J., Transport of drugs across the blood-brain barrier by nanoparticles, Curr Med Chem, 2:241-249, 2002.
Dehouck, B., Fenart, C., Dehouck, M.P., Pierce, A., Torpier, G. and Cecchelli, R., A new function for thr LDL receptor: Transcytosis of LDL across the blood-brain barrier. J Cell Biol, 138:887-889, 1997.
Luck, M., Plasma protein adsorption als Moglicher Schlusselfaktor fur eine kontrollierte Arzneistoffapplikation mit partikularen Tragern. Ph. D. Thesis, Freie Universitat Berlin, pp 130-154, 1997.
Zordan-Nudo, T., Ling, V., Liv, Z. and Georges, E., Effect of nonionic detergents on P-glycoprotein drug binding and reversal of multidrug resistance. Cancer Res, 53:5994-6000, 1993.
Olivier, J-C., Fenart, L., Chauvet, R., Pariat, C., Cecchelli, R., and Couet, W., Indirect evidence that drug brain targeting using polysorbate-80 coated polybutylcyanoacrylate nanoparticles is related to toxicity, Pharm Res, 16:1836-1842, 1999.
Neuwelt, E.A. and Dahlborg, S.A., Bloodbrain barrier disruption in the treatment of brain tumors: clinical implications, in Neuwelt EA (ed), Implications of the Blood Brain Barrier and its Manipulation: Clinical Aspects. Vol. 2, Plenum Press, New York, pp 195262, 1989.
Neuwelt, E.A., Wiliams, P.C., Mickey, B.E., Frenkel, E.P. and Henner, W.D., Therapeutic dilemma of disseminated CNS minoma and the potential of increased platinum-based chemotherapy delivery with osmotic bloodbrain barrier disruption. Pediatr Neurosurg 21:1622, 1994.
Doran, S.E., Ren, X.D., Betz, A.L., Pagel, M.A., Neuwelt, E.A., Roessler, B.J. and Davidson, B.L., Gene expression from recombinant viral vectors in the central nervous system after bloodbrain barrier disruption. Neurosurgery, 36:965970, 1995.
Neuwelt, E.A., Weissleder, R., Nilaver, G., Kroll, R.A., Roman-Goldstein, S., Szumowski, J., Pagel, M.A., Jones, R.S., Remsen, L.G. and McCormick, C.I., Delivery of virus-sized iron oxide particles to rodent CNS neurons. Neurosurgery, 34:777784, 1994.
Hiesiger, E.M., Voorhies, R.M., Basler, G.A., Lipschutz, L.E., Posner, and Shapiro, W.R., Opening the bloodbrain and bloodtumor barriers in experimental rat brain tumors: the effect of intracarotid hyperosmolar mannitol on capillary permeability and blood flow. Ann Neurol, 19:5059, 1986.
Dahlborg, S.A., Henner, W.D., Crossen, J.R., Tableman, M., Petrillo, A., Braziel, R. and Neuwelt, E.A., Non-AIDS primary CNS lymphoma: first example of a durable response in a primary brain tumor using enhanced chemotherapy delivery without phosphoro-cognitive loss and without radiotherapy. Cancer J Sci Am, 2:166174, 1996.
Kaya, M., Chang, L., Truong, A. and Brightman, M.W., Chemical induction of fenestrae in vessels of the bloodbrain barrier. Exp Neurol, 142:613, 1996.
Neuwelt, E.A., Barnett, P.A., Hellstrom, K.E., Hell strom, I., McCormick, C.I. and Ramsey, F.L., Effect of blood-brain barrier disruption on intact and fragmented monoclonal antibody localization in intracerebral lung carcinoma xenografts. J Nucl Med, 35:1831-1841, 1994.
Kroll, R.A. and Neuwelt, E.A., Outwitting the blood-brain barrier for therapeutic purposes: osmotic opening and other means, Neurosurgery, 42:1083-1099, 1998.
Rapoport, S.I., Osmotic opening of blood-brain barrier: principles, mechanism and therapeutic applications, Cell Mol Neurobiol, 20:217-230, 2000.
Miller, G., Breaking down barriers, Science, 297:1116-1118, 2002.
Chio, C.C., Baba, T. and Black, K.L., Selective bloodtumor pro-barrier disruption by leukotrienes. J Neurosurg, 77:407410, 1992.
Black, K.L., Baba, T. and Pardridge, W.M., Enzymatic barrier protects brain capillaries from leukotriene C4. J Neurosurg, 81(5):745751, 1994.
Matsukado, K., Inamura, T., Nakano, S., Fukui, M., Bartus, R.T. and Black, K.L., Enhanced tumor uptake of carboplatin and survival in glioma-bearing rats by intracarotid infusion of the bradykinin analog, RMP-7. Neurosurgery, 39:125133, 1996.
Abbott, N.J. and Romero, I.A., Transporting therapeutics across the bloodbrain barrier. Mol Med Today, 2:106113, 1996.
Emerich, D.F., Dean, R.L., and Osborn, C., Bartus, R. T., The development of the bradykinin agonist labradimil as a means to increase the permeability of the blood-brain barrier: from concept to clinical evaluation, Clin Pharmacokinet, 40:105-123, 2001.
Buchwald, P. and Bodor, N., A simple, predictive, structure-based skin permeability model, J Pharm Pharmacol, 53:1087-1098, 2001.
Krewson, C.E., Klarman, M.L. and Saltzman, W.M., Distribution of nerve growth factor following direct delivery to brain interstitium, Brain Res, 680:196-206, 1995.
Newcomb, R., Abbruscato, T.J., Singh, T., Nadasdi, L., Davis, T.P., and Miljanich, G., Bioavailability of Ziconotide in brain: influx from blood, stability and diffusion, Peptides, 21:491-501, 2000.
Harbaugh, R.E., Saunders, R. L. and Reeder, R.F., Use of implantable pumps for central nervous system drug infusions to treat neurological disease. Neurosurgery, 23(6):693-698, 1988.
Blasberg, R.G., Patlak, C. and Fenstermacher, J.D., Intrathecal chemotherapy: Brain tissue profiles after ventriculocisternal perfusion. J Pharmacol Exp Ther, 195:73-83, 1975.
Huang, T.Y., Arita, N., Hayakawa, T., and Ushio, Y., ACNU, MTX and 5-FU penetration of rat brain tissue and tumors, J Neurooncol, 45:9-17, 1999.
Thorne, R.G., Emory, C.R., Ala, T.A. and Fery, W.H., Quantitative analysis of the olfactory pathway for drug delivery to the brain. Brain Res, 692(1-2):278-282, 1995.
Born, j., Lange, T. and Kern, W., Sniffing neuropeptides: a transnasal approach to the human brain, Nat Neurosci, 5:514-516, 2002.
Illum, L., Nasal drug delivery: new development strategies, Drug Discov Today, 7:1184-1189, 2002.
Bobo, R.H., Laske, D.W., Akbasak, A., Morrison, P.F., Dedrick, R.L. and Oldfield, E.H., Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci U.S.A., 91:2076-2082, 1994.
Neuwelt, E.A., Kroll, R.A., Pagel, M.A., Muldoon, L.L and Roman Goldstein, S., Increasing volume of distribution to the brain with interstitial infusion: dose, rather than convection, might be the most important factor. Neurosurgery, 38(4):1129-1145, 1996.
Yaksh, T.L., Provencher, J.C., Rathbun, M.L., Myers, R.R., Powell, H., Richter, P. and Kohn, F.R., Safety assessment of encapsulated morphine delivered epidurally in a sustained-release multivesicular liposome preparation in dogs. Drug Deliv, 7(1):7-36, 2000.
Brem, H. and Langer, R., Polymer based drug delivery to the brain. Sci Med, 3(4):1-11, 1996.
Brem, H. and Gabikian, P., Biodegradable polymer implants to treat brain tumors, J Control Release, 74:63-67, 2001.
Fung, L.K., Ewend, M.G., Sills, A., Sipos, E.P., Thompson, R., Watts, M., Colvin, O.M., Brem, H. and Saltzman, W.M., Pharmacokinetics of interstitial delivery of carmustine, 4-hydroperoxycyclophosphamide and paclitaxel from a biodegradable polymer implant in the monkey brain. Cancer Res, 58(4):672-684, 1998.
Dang, W., Colvin, O.M., Brem, H. and Saltzman, W.M., Covalent coupling of methotrexate dextran enhances the penetration of cytotoxicity into a tissue like matrix. Cancer Res, 54:1729-1735, 1994.
Hanes, J., Batycky, R.P., Langer, R. and Edwards, D.A., A theoretical model of erosion and macromolecular drug release from biodegrading microspheres. J Pharm Sci, 86(12):1464-1477, 1997.
Jean-Pierre, B., Nathalie, F., Marie-Claire, V.J. and Philippe, M., Development of Microspheres for neurological disorders: From basics to clinical applications. J Controlled Release, 65(1-2):285-296, 2000.
Couvreur, P., Dubernet, C. and Puisieux, F., Controlled drug delivery with nanoparticles: Current possidilities and future trends. Eur J Pharm Biopharm, 41:2-13, 1995.
Pilar, C., Bruno, G., Helene, C., Didier, D., Jean, A., Jene-Pierre, N., Dominique, G., Elias, F., Jean, A.P. and Patrick, C., Long-circulating PEGylated polycyanoacrylate nanoparticles as new drug carrier for brain delivery. Pharm Res, 18(8):1157-1166, 2001.
Joerg, D., Frank, M.P., Bernhard, S.A. and Ulrike, S., Influence of nanoparticles on the brain-to-serum distribution and the metabolism of valproic acid in mice. J Pharm Pharmacol, 562:1043-1047, 2000.
Leigh, K., Elisevich, K. and Rogers, K.A., Vascularization and microvascular permeability in solid versus cell-suspension embryonic neural grafts. J Neurosurg, 81(2):272-283, 1994.
Lal, B., Indurti, R.R., Couraud, P.O., Goldstein, G.W. and Laterra, J., Endothelial cell implantation and survival within experimental gliomas. Proc Natl Acad Sci U.S.A., 91(21):9695-9699, 1994.
Snyder, E.Y. and Senut, M.C., The use of nonneuronal cells for gene delivery. Neurobiol Dis, 4(2):69-102, 1997.
Yurek, D.M. and Sladek, J.R., Dopamine cell replacement: Parkinson's disease. Annu Rev Neurosci, 13:415-440, 1990.
Raffel, C., Culer, K., Kohn, D., Nelson, M., Siegel, S., Gillis, F., Link, C.J., Villablanca, J.G. and Anderson, W.F., Gene therapy for the treatment of recurrent pediatric malignant astrocytomas with in-vivo tumor transduction with the herpes simplex thymidine kinase gene/ganciclovir system. Hum Gene Ther, 5(7):863-890, 1994.
Zlokovic, B.V. and Apuzzo, M.L., Cellular and molecular neurosurgery: pathways from concept to reality--part II: vector systems and delivery methodologies for gene therapy of the central nervous system. Neurosurgery, 40(4):805-812, 1997.
Lockman, P.R., Mumper, R.J., Kahn, M.A. and Allen, D.D, Nanoparticle technology for drug delivery across the blood-brain barrier, Drug Dev Ind Pharm, 28(1):1-12, 2002.
Oyewumi, M.O. and Mumper, R.J., Gadolinium-loaded nanoparticles engineered from microemulsion templates. Drug Dev Ind Pharm, 28(3):317-28, 2002.
Rohlff, C. and Southan, C., Proteomic approaches to central nervous system disorders. Curr Opin Mol Ther, 4:251-258, 2002.
Corresponding Author: Ambikanandan Misra, Pharmacy Department, Faculty of Technology & Engineering, M.S.University of Baroda, Kalabhavan, Vadodara 390001. Gujarat. misraan@satyam.net.in, misraan@hotmail.com
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps