J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(1):33-66, 2003
Pharmaceutical approaches to colon targeted drug delivery systems.
M. K. Chourasia, S. K. Jain1
Pharmaceutics Research Projects Laboratory, Department of Pharmaceutical Sciences, Dr. Hari Singh Gour University, Sagar, IndiaReceived 18 July 2002, Revised 16 January 2003, Accepted 3 February 2003
PDF version
Abstract
PURPOSE. Although oral delivery has become a widely accepted route of administration of therapeutic drugs, the gastrointestinal tract presents several formidable barriers to drug delivery. Colonic drug delivery has gained increased importance not just for the delivery of the drugs for the treatment of local diseases associated with the colon but also for its potential for the delivery of proteins and therapeutic peptides. To achieve successful colonic delivery, a drug needs to be protected from absorption and /or the environment of the upper gastrointestinal tract (GIT) and then be abruptly released into the proximal colon, which is considered the optimum site for colon-targeted delivery of drugs. Colon targeting is naturally of value for the topical treatment of diseases of colon such as Chron's diseases, ulcerative colitis, colorectal cancer and amebiasis. Peptides, proteins, oligonucleotides and vaccines pose potential candidature for colon targeted drug delivery. METHODS. The various strategies for targeting orally administered drugs to the colon include covalent linkage of a drug with a carrier, coating with pH-sensitive polymers, formulation of timed released systems, exploitation of carriers that are degraded specifically by colonic bacteria, bioadhesive systems and osmotic controlled drug delivery systems. Various prodrugs (sulfasalazine, ipsalazine, balsalazine and olsalazine) have been developed that are aimed to deliver 5-amino salicylic acid (5-ASA) for localized chemotherapy of inflammatory bowl disease (IBD). Microbially degradable polymers especially azo crosslinked polymers have been investigated for use in targeting of drugs to colon. Certain plant polysaccharides such as amylose, inulin, pectin and guar gum remains unaffected in the presence of gastrointestinal enzymes and pave the way for the formulation of colon targeted drug delivery systems. The concept of using pH as a rigger to release a drug in the colon is based on the pH conditions that vary continuously down the gastrointestinal tract. Times dependent drug delivery systems have been developed that are based on the principle to prevent release of drug until 3-4 h after leaving the stomach. Redox sensitive polymers and bioadhesive systems have also been exploited to deliver the drugs into the colon. RESULTS. The approach that is based on the formation of prodrug involves covalent linkage between drug and carrier. The type of linkage that is formed between drug and carrier would decide the triggering mechanism for the release of drug in colon. The presence of azo reductase enzymes play pivotal role in the release of drug from azo bond prodrugs while glycosidase activity of the colonic microflora is responsible for liberation of drugs from glycosidic prodrugs. Release of drugs from azo polymer coated dosage forms is supposed to take place after reduction and thus cleavage of the azo bonds by the azoreductase enzymes present in the colonic microflora. Natural polysaccharides have been used as tools to deliver the drugs specifically to the colon. These polysaccharides remain intact in the physiological environment of stomach and small intestine but once the dosage form enters into colon, it is acted upon by polysaccharidases, which degrades the polysaccharide and releases the drug into the vicinity of bioenvironment of colon. However, they should be protected while gaining entry into stomach and small intestine due to enormous swelling and hydrophilic properties of polysaccharides. This has been achieved either by chemical crosslinking or by addition of a protective coat. Formulation coated with enteric polymers releases drug when pH move towards alkaline range while as the multicoated formulation passes the stomach, the drug is released after a lag time of 3-5 h that is equivalent to small intestinal transit time. Drug coated with a bioadhesive polymer that selectively provides adhesion to the colonic mucosa may release drug in the colon. CONCLUSIONS. Improved drug delivery systems are required for drugs currently in use to treat localized diseases of the colon. The advantages of targeting drugs specifically to the diseased colon are reduced incidence of systemic side effects, lower dose of drug, supply of the drug to the biophase only when it is required and maintenance of the drug in its intact form as close as possible to the target site.
Introduction
The oral route is considered to be most convenient for administration of drugs to patients. Oral administration of conventional dosage forms normally dissolves in the stomach fluid or intestinal fluid and absorb from these regions of the GIT depends upon the physicochemical properties of the drug. It is a serious drawback in conditions where localized delivery of the drugs in the colon is required or in conditions where a drug needs to be protected from the hostile environment of upper GIT. Dosage forms that deliver drugs into the colon rather than upper GIT proffers number of advantages. Oral delivery of drugs to the colon is valuable in the treatment of diseases of colon (ulcerative colitis, Chron's disease, carcinomas and infections) whereby high local concentration can be achieved while minimizing side effects that occur because of release of drugs in the upper GIT or unnecessary systemic absorption. The colon is rich in lymphoid tissue, uptake of antigens into the mast cells of the colonic mucosa produces rapid local production of antibodies and this helps in efficient vaccine delivery (1). The colon is attracting interest as a site where poorly absorbed drug molecule may have an improved bioavailability. This region of the colon is recognized as having a somewhat less hostile environment with less diversity and intensity of activity than the stomach and small intestine. Additionally, the colon has a longer retention time and appears highly responsive to agents that enhance the absorption of poorly absorbed drugs. Apart from retarding or targeting dosage forms, a reliable colonic drug delivery could also be an important starting position for the colonic absorption of perorally applied, undigested, unchanged and fully active peptide drugs. As the large intestine is relatively free of peptidases such special delivery systems will have a fair chance to get their drug sufficiently absorbed after peroral application. The simplest method for targeting of drugs to the colon is to obtain slower release rates or longer release periods by the application of thicker layers of conventional enteric coatings or extremely slow releasing matrices. Various pharmaceutical approaches that can be exploited for the development of colon targeted drug delivery systems are summarised in Table 1.
Table 1: Various pharmaceutical approaches to colon targeted drug delivery systems.
Covalent linkage of the drug with a carrier
It involves the formation of a covalent linkage between drug and carrier in such a manner that upon oral administration the moiety remains intact in the stomach and small intestine.
This approach chiefly involves the formation of prodrug, which is a pharmacologically inactive derivative of a parent drug molecule that requires spontaneous or enzymatic transformation in the biological environment to release the active drug. Formation of prodrugs has improved delivery properties over the parent drug molecule. The problem of stability of certain drugs from the adverse environment of the upper GIT can be eliminated by prodrug formation, which is converted into parent drug molecule once it reaches into the colon. Site specific drug delivery through site specific prodrug activation may be accomplished by the utilization of some specific property at the target site, such as altered pH or high activity of certain enzymes relative to the non-target tissues for the prodrug-drug conversion.
Azo bond conjugates
The intestinal microflora is characterized by a complex and relatively stable community of microorganism, many with physiological functions, which play vital roles in health and disease. In addition to protection of the patient against colonization of the intestinal tract by potentially pathogenic bacteria, the indigenous microflora are responsible for a wide variety of metabolic processes, including the reduction of nitro and azo groups in environmental and therapeutic compounds (2-4).
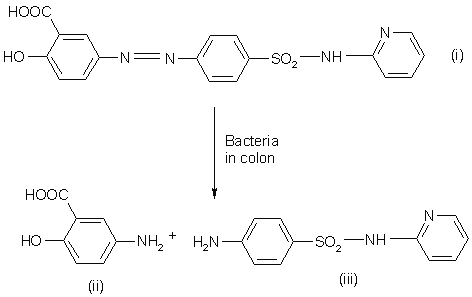
Sulphasalazine was introduced for the treatment of rheumatoid arthritis and anti-inflammatory disease. Chemically it is salicylazosulphapyridine (SASP), where sulfapyridine is linked to a salicylate radical by an azo bond (5). When taken orally, only a small proportion of the ingested dose is absorbed from the small intestine and the bulk of the sulphasalazine reaches the colon intact. There it is split at the azo bond by the colonic bacteria with the liberation of sulphapyridine (SP) and 5-ASA (Figure 1). However sulphapyridine is seems to be responsible for most of the side effects of sulphasalazine and hence various new approaches for the treatment of IBD have emerged.
Figure 1: Hydrolysis of sulfasalazine (i) into 5-aminosalicylic acid (ii) and sulfapyridine (iii).
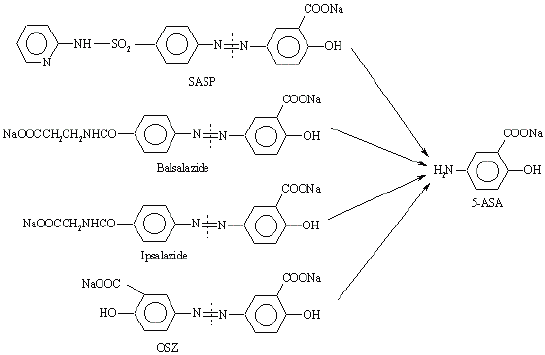
The need for less toxic carrier moieties has led to the development and testing of a number of other azo-bond prodrugs. By replacing the carrier molecule with others, a number of prodrugs of 5-ASA can be prepared e.g. p-aminohippurate (4-amino benzoyl glycine) in ipsalazine, 4-amino benzoyl-b-alanine in balsalazine (6), p-aminobenzoate in HB-313 (7) or a nonabsorbable sulphanilamide ethylene polymer in poly-ASA (8). The most interesting prodrug is olsalazine (OSZ) which is a dimer representing two molecules of 5-ASA that are linked via an azo bond. When olsalazine reaches the large intestine, it is cleaved releasing two molecules of 5-ASA for every mole of olsalazine administered. This prodrug is absorbed intact from the human GIT to only a very limited extent and, as with SASP, 5-ASA and acetyl-5-ASA are recovered in the feces following oral administration of OSZ (9, 10). It has been shown clinically that an intact GIT and a normal microflora population are required for effective splitting of OSZ (11, 12). Fecal recovery of 5-ASA has been found to be virtually identical to an equamolar dose of SASP (13). Clinical trials have been encouraging although watery diarrhea has emerged as new and troublesome side effect, which generally affects about 15% of the patients. This side effect appears to be related to a combination of gastrointestinal transit and a stimulation of small intestinal secretion (14, 15). A second azo bond prodrug developed is balsalazine, which is 5-ASA azo-linked to 4-aminobenzoyl- β -alanine (Figure 2).
Figure 2: The chemical structure of SASP, balsalazide, ipsalazide and OSZ showing the site of bacterial cleavage leading to formation of the active agent 5-ASA.
This carrier is designed to be inherently less toxic than SP while maintaining the poor absorbability of the prodrug from the upper GIT. The promoiety is only minimally absorbed following azo-reduction in the colon. Clinical trials suggest that balsalazine is useful in maintaining remission in the ulcerative colitis (16) with fewer side effects than are associated with SASP maintenance therapy. Another prodrug called ipsalazine (Figure 2) has also been synthesized and tested as a carrier for 5-ASA. Despite promising pharmacokinetic data, ipsalazine has not been developed further (17).
Polymeric prodrug with drug molecules linked directly to a high molecular weight polymeric backbone has also been investigated for colon targeted drug delivery. The linkage between the drug and polymer is susceptible to enzymatic attack in the large intestine and the drug is released at this site. In case of polymeric prodrugs, the large size of the prodrug hinders absorption from the upper GIT. Dynapol Corporation developed a compound (poly asa) that was based on the SASP carrier concept i. e. SP unit is linked to an inert polymer backbone (18, 19). The carrier is a polysulfonamidoethylene to which 5-ASA is azo linked. The mechanism of delivery of 5-ASA to the colon is essentially that of SASP i.e. reduction of azo bonds by azoreductase enzymes. Burroughs Wellcome further developed poly asa under the name BW73Y (20).
Biorecognizable HPMA copolymer conjugates for colon-specific delivery of 9-aminocamptothecin (9-AC) was designed. They hold 9-AC bound via spacers containing amino acid residues and aromatic azo bonds. In vitro release profiles of 9-AC from HPMA copolymer conjugates were evaluated under artificial conditions that simulated large intestinal azoreductase and peptidase activities. The studies indicated that the azo bond was reduced first, followed by the release of unmodified 9-AC from the 9-AC containing fragment by peptidases. Release profiles depended on the chemical structure of the peptide part of the spacer. Conjugates containing leucylalanine showed high colon-specific release of 9-AC when compared to alanine containing conjugates (21).
Glycoside conjugates
Steroid glycosides and the unique glycosidase activity of the colonic microflora form the basis of a new colon targeted drug delivery system. Drug glycosides are hydrophilic and thus, poorly absorbed from the small intestine. Once such a glycoside reaches the colon it can be cleaved by bacterial glycosidases, releasing the free drug to be absorbed by the colonic mucosa.
The major glycosidases identified in human feces are b -D-galactosidase, b -D-glucosidase, a -L-arabinofuranosidase, b -D-xylopyranosidase (22). These enzymes are located at the brush border and hence access to the substrate is relatively easy. In the plant kingdom numerous compounds are found as glycosides. Certain drugs act as glycon and can be conjugated to different sugar moieties which results in the formation of glycosides. Due to the bulky and hydrophilic nature of these glycosides, they do not penetrate the biological membrane upon ingestion (23). Various naturally occurring glycosides, e.g. the sennosides, have been used for laxative action for ages. When taken orally, intact sennosides are more efficient as laxative than sugar free aglycones. These sennosides are activated by colonic microflora to generate rhein anthones, which gives the desired laxative effect (24). Glycosidase activity of the GIT is derived from anaerobic microflora in the large bowel or the sloughed or exfoliated cells of the small intestine (25,26).
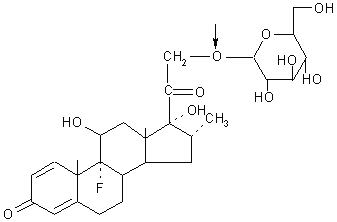
Friend and Chang (27) prepared dexamethason-21- b -glucoside (Figure 3) and prednisolone-21- b -glucoside for delivery of these steroids to the colon.
Figure 3: Dexamethasone-21-b-D-glucoside (Arrow shows site of action of glycosidase).
Hydrolysis of prodrugs by b -glucosidase and fecal homogenates in vitro released the free steroids. Glucosides were administered to rats intragastrically to determine when and where the free steroids were released. Unmodified dexamethasone and prednisolone were also given to rats intragastrically to compare absorption of the glucosides with the free steroids. Both glucosides were found to reach the rat lower intestine in 4-5 h, where they were rapidly hydrolyzed, releasing the free steroids. In vivo studies on dexamethasone- b -D-glucoside revealed that nearly 60% of an oral dose of glucoside reached the caecum whereas in case of prednisolone- b -D-glucoside, only 15% reached to the caecum. When free steroids were administered orally, they were almost absorbed in the small intestine and less than 1% of oral dose reached at the colon.
The influence of prodrug structure on specificity of glycoside/glycosidase based colon-specific drug delivery was studied by preparing nine steroid glycosides, measuring their relative lipophilicities and hydrolyzing them with bacterial glycosidases from rat intestines (28). The 21-yl b -D-glucosides and galactosides of dexamethasone, prednisolone, hydrocortisone and fludrocortisone and 21-yl b -D-cellobioside of prednisolone were prepared by a modified Koenigs-Knorr reaction. The deacetylated glycoside prodrugs along with the p-nitrophenyl derivatives of b -D-glucoside, galactoside and cellobioside were subjected to hydrolysis by the contents of the rat stomach, proximal small intestine (PSI), distal small intestine (DSI) and caecum. All the prodrugs were hydrolyzed slowly by PSI and stomach contents, more rapidly by contents of the DSI, and most rapidly by caecal contents. Furthermore, the prodrugs themselves had very different susceptibilities to hydrolysis. Hydrolysis rates catalyzed by DSI contents decreased in the following order: prednisolon-21-yl b -D-galactoside > prednisolon-21-yl b -D-glucoside > prednisolon-21-y l b -D-cellobioside > dexamthason-21-yl b -D- galactoside > dexamthason-21-yl b -D- glucoside. Hydrolysis of prednisolon-21-yl b -D-cellobioside was only half that of prednisolon-21-yl b -D-glucoside and one forth that of prednisolon-21-yl b -D-galactoside. Hydrolysis of all the products in caecal contents was rapid, with the exceptions of hydrocortison-21-yl b -D-glucoside and fludrocortison-21-yl b -D-glucoside, which were hydrolyzed more slowly than the other glucoside prodrugs. Eadie-Hofstee plots for hydrolysis of the glucoside compounds suggested that bacterial b -D-glucosidase activity in the colon might be more heterogeneous in nature than b -D-galactosidase activity.
In vitro studies performed specifically on dexamethason- b -D-glucoside (29-31) revealed that both GIT tissues and GIT contents of guinea pig showed b -glucosidase activity. Among the tissues maximum activity was seen in tissues of PSI whereas among the contents maximum activity was seen in the caecum and the colon. For in vivo studies experimental IBD was induced using degraded carrageenan in guinea pig. 0.65 mol/kg dexamethason- b -D-glucoside was equally effective as 1.3 mol/kg of dexamethason alone in reducing the total number of ulcers. The results indicated that a lower dose of dexamethasone, administered, as its glucoside prodrug could be equally efficacious relative to higher dose of dexamethasone.
Glucuronide conjugates
Glucuronide and sulphate conjugation is the major mechanisms for the inactivation and preparation for clearance of a variety of drugs. Bacteria of the lower GIT, however, secrete b -glucuronidase and can deglucuronidate a variety of drugs in the intestine (32). Since the deglucuronidation process results in the release of active drug and enables its reabsorption, glucuronide prodrugs would be expected to be superior for colon targeted drug delivery.
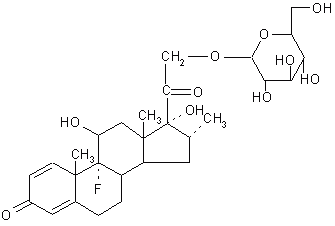
Morphine-dependent rats were used to evaluate the effects of the narcotic antagonists, naloxone and nalmefene, and their glucuronide conjugates on the gastrointestinal tract and various parameters of brain-mediated withdrawal. When administered subcutaneously nalmefene hydrochloride caused a dose-dependent tail skin temperature increase, whereas nalmefene glucuronide was ineffective. Nalmefene precipitated brain-mediated morphine withdrawal at doses as low as 10 mg/kg, whereas nalmefene glucuronide was ineffective at doses as high as 1 mg/kg. After per oral administration of the drugs, naloxone hydrochloride and nalmefene hydrochloride caused diarrhea, withdrawal behavior and tail skin temperature responses by 15 minutes. In contrast, after per oral administration of the glucuronide conjugate of either narcotic antagonist, diarrhea was delayed for 75 to 203 minutes. This latency probably reflects the required transit time to the lower gastrointestinal tract. About 0.2 to 0.5% of the dose of the narcotic antagonist administered orally as the glucuronide was absorbed systemically. These results indicate that per oral administration of the glucuronide conjugates of naloxone and nalmefene results in delivery of the narcotic antagonists to the colon (33). Haeberlin et al. (34) prepared a dexamethasone- b -D-glucuronide prodrug (Figure 4).
Figure 4: Dexamethasone- b -D-glucuronide.
The authors conducted a study on conventional colitic and germ free rats and demonstrated that there was a 30-fold increase in luminal b -D-glucuronidase activity between DSI and caecum in normal rats. The treatment of ulcerative colitis was improved by the synthesis of budenoside and dexamethasone conjugates of glucuronic acid and dextran (35, 36). The system showed excellent performance in ulcerative colitis and reduces the systemic toxicity of corticosteroids including adrenal suppression by immurement of activity of the drug in large intestine. The preclinical efficacy of dexamethasone- b -D-glucuronide was confirmed in the rat model of ulcerative colitis and Chron's colitis (37). Nolen et al. (38) investigated the steady-state pharmacokinetics of corticosteroid delivery from glucuronide prodrugs in normal and colitic rats. Two prodrugs, dexamethasone- b -D-glucuronide (DXglrd) and Budesonide- b -D-glucuronide (BUDglrd) were administered by intragastric infusion to conventional and colitic rats. In addition, dexamethasone and Budesonide were administered either intragastrically or subcutaneously to healthy and colitic rats and colon-specific delivery was assessed using the drug delivery index. In conventional rats, drug delivery indices for DXglrd ranged from about five to as high as 11 in the luminal contents relative to dexamethasone administered subcutaneously or intragastrically. Drug delivery index values were also elevated in the mucosa of both healthy and colitic rats following intragastric administration of DXglrd. Budesonide was delivered somewhat less effectively from BUDglrd to the rat large intestine than was dexamethasone from DXglrd.
Cyclodextrin conjugates
Cyclodextrins (CyDs) are cyclic oligosaccharides consisted of six to eight glucose units through a -1,4 glucosidic bonds and have been utilized to improve certain properties of drugs such as solubility, stability and bioavailability. The interior of these molecules is relatively lipophilic and the exterior relatively hydrophilic, they tend to form inclusion complexes with various drug molecules (39-43). They are known to be barely capable of being hydrolyzed and only slightly absorbed in passage through the stomach and small intestine; however, they are fermented by colonic microflora into small saccharides and thus absorbed in the large intestine (44-46). Because of their bioadaptability and multi-functional characteristics, CyDs are capable of alleviating the undesirable properties of drug molecules in various routes of administration through the formation of inclusion complexes. In an oral drug delivery system, the hydrophilic and ionizable CyDs can serve as potent drug carriers in the immediate release and delayed release-formulations, respectively, while hydrophobic CyDs can retard the release rate of water-soluble drugs. Since CyDs are able to extend the function of pharmaceutical additives, the combination of molecular encapsulation with other carrier materials will become effective and a valuable tool in the improvement of drug formulation. Moreover, the most desirable attribute for the drug carrier is its ability to deliver a drug to a targeted site; conjugates of a drug with CyDs can be a versatile means of constructing a new class of colon targeting prodrugs.
It has been proved through a study in healthy human volunteers that b CyDs are meagerly digested in small intestine but are completely degraded by the microflora of the colon. Most bacterial strains that are isolated from human being are capable of degrading CyDs. It has been proved by their ability to grow on cyclodextrins by utilizing them as the sole carbon source and by the stimulation of cyclodextrinase activity by as low as 2-4 h of exposure to cyclodextrins. This property of the drug may be exploited for the formation of colon targeted drug delivery systems. Several CyD conjugates have been prepared and the enantioselective hydrolysis has described (47-49).
An anti-inflammatory drug biphenylacetic acid (BPAA) as model drug was selectively conjugated onto one of the primary hydroxyl groups of a -, b - and γ- CyDs through an ester or amide linkage, and the in vivo drug release behavior of these prodrugs in rat gastrointestinal tract after oral administration was investigated. The CyD prodrugs were stable in rat stomach and small intestine and negligibly absorbed from these tracts. Three to six h after oral administration, most of the prodrugs had moved to the caecum and colon. The a- and γ-CyD amide prodrugs were hydrolyzed to the maltose conjugate in the caecum and colon, and these prodrugs and the conjugates were negligibly absorbed. On the other hand, the a-and γ-CyD ester prodrugs produced BPAA in the caecum and colon, and BPAA appeared in the blood after 3-6 h. Both b-CyD amide and ester prodrugs released only small or negligible amounts of the maltose conjugate or BPAA in the caecum and colon within 24 h, probably due to the low solubility in the biological media. Further, the anti-inflammatory effect of the γ-CyD ester prodrug was evaluated using the model of carageenan-induced acute edema in rat paw and compared with those of BPAA alone and the BPAA/b-CyD complex prepared by the kneading method in a molar ratio of 1:1. In the case of b-CyD complex, a rapid anti-inflammatory response was observed from the small intestine after a fast dissolution of the complex. In sharp contrast, the γ-CyD ester prodrug required a fairly long lag time to exhibit the drug activity, because BPAA was produced after the prodrug had reached the caecum and colon. These results clearly suggest that the CyD prodrug approach could provide a versatile means for constructions of not only colon-specific delivery systems but also delayed-release system of certain drugs (50, 51).
Hiramaya et al. (52) prepared two CyD conjugates where one primary hydroxyl group of b-CyDs was substituted by BPAA through an ester or amide linkage. Aqueous solubility of the conjugates was lower than those of other drug or parent compound. The amide conjugate was stable in aqueous solution and in rat biological fluids and gastrointestinal contents. The ester conjugate released the drug preferentially when incubated with the contents of caecum or colon, whereas no appreciable drug release was observed on incubation with the contents of either stomach or intestine in intestinal or liver homogenates or in rat blood. Prednisolone, a typical glucocorticoid, has been widely used for the treatment of IBD. However, when Prednisolone is administered orally, a large amount of the drug is absorbed from the upper GIT and causes systemic side effects. The anti-inflammatory effect and systemic side effect of the prednisolone succinate/alpha-cyclodextrin ester conjugate after oral administration were studied using IBD model rats. The systemic side effect of the conjugate was much lower than that of prednisolone alone when administered orally. The lower side effect of the conjugate was attributable to passage of the conjugate through the stomach and small intestine without significant degradation or absorption, followed by the degradation of the conjugate site-specifically in the large intestine (53). Yano et al. (54) studied the antiinflammatory and systemic side effects of the prednisolone-appended a-CyD prodrug after oral administration to the 2,4,6-trinitrobenezenusulfonic acid induced colitis rats. Prednisolone was introduced at on the secondary hydroxyl groups of a, b- and γ- CyDs. The intracolonic administration of the prednisolone-appended a-CyD prodrug to colitis rats significantly alleviated the side effect of the drug, while maintaining the therapeutic activity.
Two of the parent CyDs, a-CyD and b-CyDs are known to be parenterally unsafe due to severe nephrotoxicity (55) but both a- and b-CyDs has been used orally in food products in various approved pharmaceuticals. The etiology of nephrotoxicity of both a- and b- CyDs is unknown but appears to be related to either CyD uptake by the kidney tubule cells followed by disruption of intracellular function, or the extraction of lipid membrane components by the CyDs (56). Modification of the parent CyDs to improve safety while maintaining the inherent properties has been the goal of various scientists. Many modified CyDs have been prepared for the delivery of bioactive agents to the colon. When the modified CyDs are administered orally, their absorption is low and most of them are excreted intact in the feces, as demonstrated by epichlorhydrin DM-b-CyDs (147 M) and HP-b-CyD in rats, dogs and humans (57, 58). Hydrophobic CyD may be useful in various controlled release formulations of water-soluble drugs including peptides and protein drugs (59).
Dextran conjugates
Dextran ester prodrug was prepared and in vitro release revealed that release of naproxan from prodrug was several folds higher in caecum homogenates than in control medium or homogenates of the small intestine of pig (60, 61). The bioavailability of naproxan after oral administration of a dextran T-70-naproxan ester prodrug in pigs was assessed by Harboe et al. (62). Compared to the administration of an oral solution of an equivalent dose of naproxan the average absorption fraction for the conjugate amounted to 91%. It was established that several features of the prodrug indicated that naproxan was released from the prodrug prior to systemic absorption and that drug activation involved the action of one or more enzyme systems located in the gastrointestinal tract. It was observed in rabbits, the plasma concentration-time curves for the conjugate were characterized by an initial lag time of about 2-3 h, whereas naproxan was detected in plasma immediately after per oral administration of the drug compound per se. The distribution of the prodrug along the GIT at various times after conjugate administration was assessed qualitatively by HPLC analysis of conjugated and free naproxan in various segments of the GIT. From these experiments it was suggested that drug regeneration was effective in the bowel below the ileum.
Dextran ester prodrugs of metronidazole have been prepared and characterized (63-65). Mcleod et al. (66-68) synthesized dextran ester prodrugs of dexamethasone and methylprednisolone and proved the efficacy of the prodrugs for delivering drugs to the colon. In this study, methyl prednisolone and dexamethasone were covalently attached to dextran by the use of a succinate linker. In addition, dexamethasone was attached by glutaric acid to investigate the effect of linker molecule on hydrolysis kinetics. The kinetics of degradation of the hemiester and corresponding dextran conjugates were measured as a function of pH and temperature. Intermolecular migration of the linker molecule from the 21- to the 17-position on the glucocorticoid occurred in all three hemiester, although to a greater extent in methylprednisolone-hemiester. The dextran conjugates were also incubated at 37°C; pH 6.8 and the chemical degradation half-lives were determined. Dexamthasone-21-hemisuccinate showed half life of 75 h, dexamethasone-glutarate-dextran exhibited half life of 103 h, while methylprednisolone-succinate-dextran showed half life of 82 h.
Glucocorticoids remain the foundation of therapy for acute ulcerative colitis despite systemic side effects that limit their use. Prodrugs that selectively deliver glucocorticoids to the colon may lower the required dose and side effects.
Amino-acid conjugates
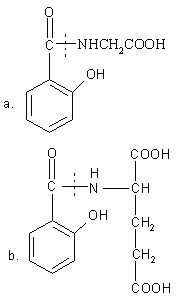
Due to the hydrophilic nature of polar groups like -NH2 and -COOH, that is present in the proteins and their basic units (i.e. the amino acids), they reduce the membrane permeability of amino acids and proteins. Various prodrugs have been prepared by the conjugation of drug molecules to these polar amino acids (69-72). Non-essential amino acids such as tyrosine, glycine, methionine and glutamic acid were conjugated to SA. The salicyluric acid (the glycine conjugate of SA) was found to be metabolized to SA by the microorganisms of the intestinal flora of rabbit and dog (Figure 5a). The prodrug was absorbed into the systemic circulation from the upper GIT and hence it was proved unsuitable for delivery of drugs to the colon. By increasing the hydrophilicity and chain length of the carrier amino acid and decreasing the membrane permeability of conjugate Nakamura et al. prepared salicylic glutamic acid conjugates (Figure 5b). This conjugate showed splendid results with minimal absorption and degradation in the upper GIT and proved suitable for colon targeted delivery of SA.
Figure 5: Glycine and glutamic acid conjugates of salicylic acid. (a) Salicyluric acid. (b) Salicyl-glutamic acid conjugate (Dotted line shows the site of cleavage).
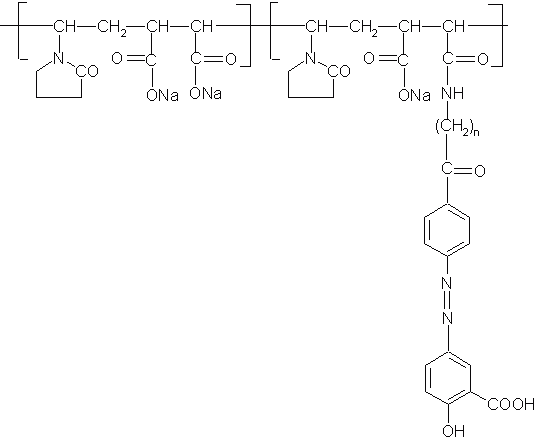
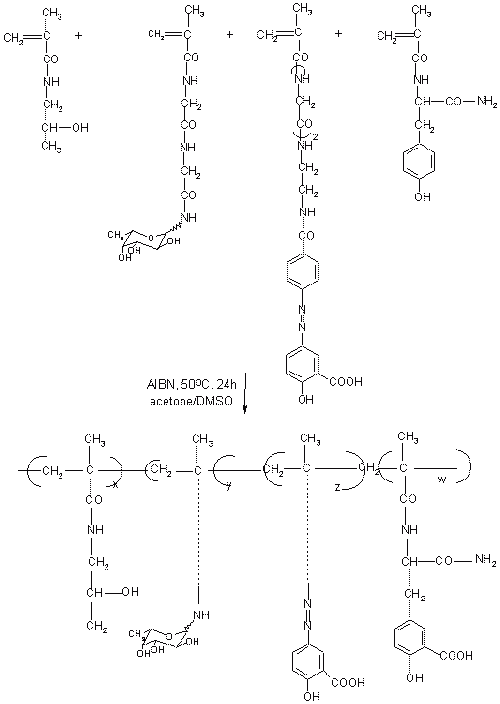
Polymeric prodrugs have developed using a spacer coupling 5-ASA via 5-amino function by an azo bond. The spacer 5-ASA conjugates is then covalently linked to poly (methyl vinyl ether/co-maleic anhydride) and poly (1-vinyl-2-pyrrolidone co-maleic anhydride) (Figure 6) and also to chloroformate-activate derivatives of dextran and poly [(2-hydroxyethyl) aspartamine]. The release of 5-ASA from polymeric prodrugs was depended upon the structure of the polymeric backbone (74). Despite the fact that all these polymeric prodrugs can deliver 5-ASA successfully to the large intestine, 5-ASA may not be the drug of choice for these systems. Indeed, the required dose of 5-ASA ranges from 0.5 to 3g daily, and since the drug makes less than 10% of the total weight of the prodrug; a very large amount would need to be taken orally.
Figure 6: Polymeric prodrug containing 5-ASA conjugate covalently linked to poly (methyl vinyl ether/co-maleic anhydride) and poly (1-vinyl-2-pyrrolidone co-maleic anhydride).
Poly-(L-aspartic acid) has been investigated as carrier for colon targeted delivery of dexamethasone (75, 76). The ester prodrug with 10% w/w drug loading was synthesized using dicyclohexyl carbodiimide as dehydrating agent in Dimethyl formamide. On the basis of in vitro studies it was concluded that maximum hydrolytic activity for this prodrug was observed in caecum and colonic contents of rats.
Approaches to deliver the intact molecule to the colon
Coating with polymers
The intact molecule can be delivered to the colon without absorbing at the upper part of the intestine by coating of the drug molecule with the suitable polymers, which degrade only in the colon.
Coating with pH-sensitive polymers
The pH-dependent systems exploit the generally accepted view that pH of the human GIT increases progressively from the stomach (pH 1-2 which increases to 4 during digestion), small intestine (pH 6-7) at the site of digestion and it increases to 7-8 in the distal ileum. The coating of pH-sensitive polymers to the tablets, capsules or pellets provide delayed release and protect the active drug from gastric fluid. The polymers used for colon targeting, however, should be able to withstand the lower pH values of the stomach and of the proximal part of the small intestine and also be able to disintegrate at the neutral of slightly alkaline pH of the terminal ileum and preferably at the ileocecal junction. These processes distribute the drug throughout the large intestine and improve the potential of colon targeted delivery systems. While this release pattern can be studied in vitro , there is no real substitute for confirming reliable performance in vivo in man. The technique of gamma scintigraphy has become the most popular method to investigate the gastrointestinal performance of pharmaceutical dosage forms (77). The threshold pH commonly employed pH-sensitive polymers are depicted in Table 2.
Table 2: Threshold pH of commonly used polymers.
The majority of enteric and colon targeted delivery systems are based on the coating of tablets or pellets, which are filled into conventional hard gelatin capsules. However, during the early stage of drug development some new chemical entities (NCE's) present a challenge in testing for efficacy due to instability in gastric fluids because of irritation in the GIT. The limited amount of drug substance available during the early stage often precludes the development of a coated pellet or tablet formulation. Since the coating process is independent of the capsule contents, there are clear advantages resulting from the ability to coat a capsule. Thus, the oral pharmacological and or therapeutic efficacy of the NCE can be determined without resorting to extensive, time consuming and in many instances, impossible at this point in the development of the NCE.
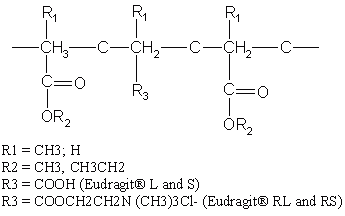
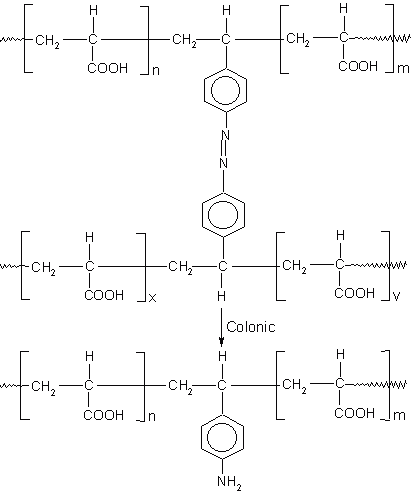
The GI residence time of the dosage forms is another important parameter for pH-dependent colon targeted drug delivery systems which is influenced by many physiological and other factors (78, 79); nevertheless, there are some generally accepted GI residence values for various parts of the GIT (80). Most commonly used pH-dependent coating polymers are methacrylic acid copolymers, commonly known as Eudragit® S (Registered trademark of Rohm Pharmaceuticals, Darmstadt, Germany), more specifically Eudragit® L and Eudragit® S (Figure 7). Eudragit® L100 and S 100 are copolymers of methacrylic acid and methyl methacrylate. The ratio of carboxyl to ester groups is approximately 1:1 in Eudragit® L100 and 1:2 in Eudragit® S 100. The polymers form salts and dissolve above pH 5.5 and disperse in water to form latex and thus avoid the use of organic solvents in the coating process. Eudragit® L30D-55 is a ready to use aqueous dispersion of Eudragit® L100-55. The water solubility of the Eudragit® S depends on the ratio of free carboxyl groups to the esterifies groups. The critical factor that influences the performance of these polymers is the pH value at which dissolution occurs. Polymers with ionizable phthalic acid groups dissolve much faster and at a lower pH than those with acrylic or methacrylic acid groups. The presence of plasticizer (81) and the nature of the salt (82, 83) in the dissolution medium also influence the dissolution rate of Eudragit®. In addition, the permeability of the film formed may depend on the type of solvent used to dissolve Eudragit® (84).
Figure 7: Chemical structures of various formulations of Eudragit®
Colon targeted drug delivery systems based on methacrylic resins has described for insulin (85), prednisolone (86), quinolones (87), salsalazine (88-92), cyclosporine (93), beclomethasone dipropionate (94) and naproxane (95). Khan et al. (96) prepared lactose-based placebo tablets and coated using various combinations of two methacrylic acid polymers, Eudragit® L100-55 and Eudragit® S100 by spraying from aqueous systems. The Eudragit® L100-55 and Eudragit® S100 combinations studied were 1:0, 4:1, 3:2, 1:1, 2:3, 1:4, 1:5 and 0:1. The coated tablets were tested in vitro for their suitability for pH dependent colon targeted oral drug delivery. The same coating formulations were then applied on tablets containing mesalazine as a model drug and evaluated for in vitro dissolution rates under various conditions. The disintegration data obtained for the placebo tablets demonstrate that disintegration rate of the studied tablets is depends on the polymer combinations used to coat the tablets, pH of the disintegration media and the coating level of the tablets. Dissolution studies performed on the mesalazine tablets further confirmed that the release profiles of the drug could be manipulated by changing the Eudragit® L100-55 and Eudragit® S100 ratios within the pH range of 5.5 to 7.0 in which the individual polymers are soluble respectively, and a coating formulation consisting of a combination of the two copolymers can overcome the issue of high GI pH variability among individuals. The results also demonstrated that a combination of Eudragit® L100-55 and Eudragit® S100 could be successfully used from aqueous system to coat tablets for colon targeted drug delivery of drugs and the formulation can be adjusted to deliver drug at any other desirable site of the intestinal region of the GIT on the basis of pH variability. Lorenzo-Lamosa et al. (97) prepared and demonstrated the efficacy of a system, which combines specific biodegradability and pH dependent release behavior. The system consists of chitosan microcores entrapped within acrylic microspheres containing diclofenac sodium as model drug. The drug was efficiently entrapped within the chitosan microcores using spray drying and then microencapsulated into Eudragit® L-100 and Eudragit® S-100 using an oil-in-oil solvent evaporation method. Release of the drug from chitosan multireservoir system was adjusted by changing the chitosan molecular weight or the type of chitosan salt. Furthermore, by coating the chitosan microcores with Eudragit®, perfect pH-dependent release profiles were attained. Numerous Eudragit® coated oral dosage forms of salsalazine are currently in use for the treatment of ulcerative colitis and Chron's disease (98-104). Morishita et al. (105) compared the insulin delivery of two formulations containing Eudragit® L-100 and Eudragit® LS respectively. Formulation containing Eudragit® S showed optimal delivery of insulin in the ileum at pH 7.
The enteric coating of HPMC capsules containing paracetamol was investigated by Cole et al. (106). Two enteric polymers Eudragit® L-30 D-55 and Eudragit® FS 30 D were studied, which were designed to achieve enteric properties and colonic release respectively. Dissolution studies demonstrated that capsules coated with Eudragit® L-30 D-55 were gastro resistant for 3 h at pH 1.2 and capsules coated with Eudragit® FS 30 D were resistant for a further 1 h at pH 6.8. Several methacrylated derivatives of Eudragit® S with different degrees of substitution were prepared for evaluation as potential coatings for colon targeted drug delivery. Water vapor transmission, in vitro dissolution and stability in function of pH were investigated using the technique of isolated films. The data presented demonstrate relative differences in physico-chemical characteristics due to differences in degree of substitution (107). Ondansetorn and Budesonide drugs, which are used for local treatment of intestinal disorders, were efficiently entrapped in a new microparticulate system, which combines pH-dependent and controlled drug release properties. This system was formulated by drug loaded cellulose acetate butyrate (CAB) microspheres coated by an enteric polymer (Eudragit® S). Both, CAB cores and pH-sensitive microcapsules were prepared by the emulsion-solvent evaporation technique. The in vitro drug release studies of pH-sensitive microcapsules containing the hydrophobic cores showed that no drug was released below pH 7. After that, CAB microspheres efficiently controlled the release of Budesonide, the release behavior being affected by the different polymer concentration used in their preparation (108). The disadvantage of this approach is the lack of consistency in the dissolution of the polymer at the desired site. Depending on the intensity of the GI motility, the dissolution of the polymer can be complete deep in the colon or at the end of the ileum. Moreover, many factors such as the presence of short chain fatty acids, residues of bile acids, carbon dioxide or other fermentation products can reduce the colonic pH to approximately 6 and call its pH as a trigger into question.
Ashford et al. (109) have shown that pH-sensitive polymers are not suitable for colon targeted drug delivery systems due to poor site specificity. The long lag times at the ileocaecal junction and fast transit indicate that a single unit may not be the best dosage form for a colon targeted drug delivery system. The late disintegration of a single-unit dosage form creates a particular problem due to the abnormal release of contents and will results in loss of much of the opportunity for local action or absorption in the proximal colon. This problem can be partially rectified by targeting multiple-unit dosage forms.
The microparticulate system consisted of Budesonide-containing hydrophobic cores, microencapsulated within an enteric polymer was prepared by Rodriguez et al. (110), which solubilizes at above pH 7, thus combining pH-sensitive and controlled-release properties. Colonic injury and inflammation were assessed by measuring colon/body weight ratio, myeloperoxidase activity, and by scoring macroscopic and histological damage in colitic rats. Rats were treated orally with Budesonide, included in the developed system, once a day for 4 days after the induction of inflammation. A Budesonide suspension and Budesonide-containing enteric microparticles were included as control formulations in the experimental design. The administration of the new Budesonide delivery system significantly reduced the colon/body weight ratio compared with the administration of control formulations. Similarly, myeloperoxidase activity and macroscopic and histological damage of the inflamed colonic segments decreased significantly when the Budesonide formulation was administered compared with the results obtained after oral administration of the drug suspension. There were no significant differences, however, when the new treatment was compared with the control formulation consisting of simple enteric microparticles.
Markus et al. (111) developed a multi-unit dosage form containing 5-ASA for the treatment of ulcerative colitis. Pellets were prepared by a granulation and spheronization process and then coated with a new pH sensitive poly(meth)acrylate copolymer (Eudragit® FS 30D) to achieve site specific drug release close to the ileocaecal valve. From the dissolution studies it was concluded that pellets released rapidly at pH values above 7.5. Between 6.8 and 7.2 drug release was found to be zero order, while at pH 6.5 and below no release occurred. In a biorelevant medium, which simulates the fasting proximal small intestine fluid, it was shown that neither surfactants (sodium taurocholate and lecithin) nor changes in ionic strength trigger drug release. Compared to 5-ASA pellets coated with the well established Eudragit® S, and to currently marketed products licensed for the treatment of ulcerative colitis, the multi-unit dosage form coated with the new polymer exhibited an in vitro dissolution profile more appropriate to the pH profile of the ileum and the colon observed in ulcerative colitis patients.
Coating with biodegradable polymers
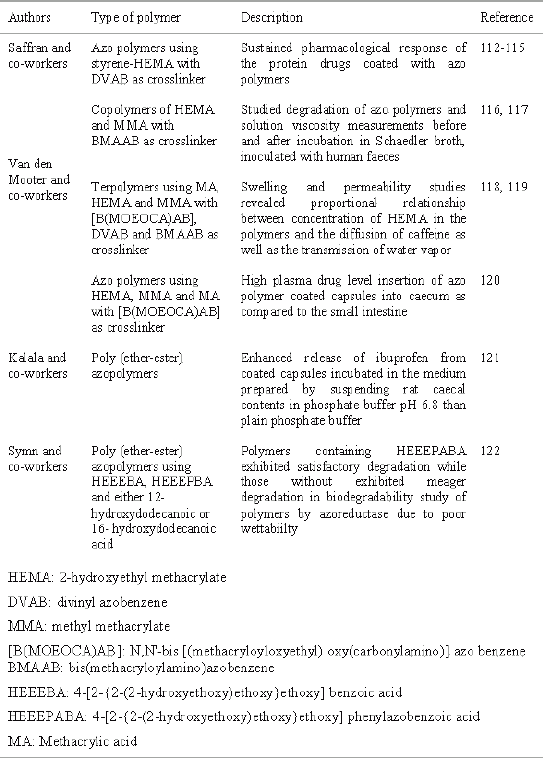
The bioenvironment inside the human GIT is characterized by the presence of complex microflora especially the colon that is rich in microorganisms that are involved in the process of reduction of dietary component or other materials. Drugs that are coated with the polymers, which are showing degradability due to the influence of colonic microorganisms, can be exploited in designing drugs for colon targeting. These bacterial degradable polymers especially azo polymers have been explored in order to release an orally administered drug in the colon. Actually, upon passage of the dosage form through the GIT, it remains intact in the stomach and small intestine where very little microbially degradable activity is present that is quiet insufficient for cleavage of polymer coating. Release of the drugs from azo polymer coated formulation is supposed to take place after reduction and thus degradation of the azo bonds by the azo reductase enzymes released by the azo bacters present in the colonic microflora. Since the concept of this strategy is based on the metabolic activity of azo reductase produced by azo bacters of colon, the bacterial degradation of polymeric coating may be effected by several other factors e.g. dietary fermentation precursors, type of food consumed and coadministration of chemotherapeutic agents. Administration of antibiotics may result in the partial or complete destruction of colonic microflora, which adversely affect the release of bioactive agents. Table 3 summarizes the synthesis and description of various azo polymers that have been exploited for colon targeted drug delivery.
Table 3: Azo polymers for colon targeted drug delivery.
Linear-type-segmented polyurethanes containing an azo group in the main chain have been synthesised as coating material (123-125). Since this polymer was degraded specifically by the action of intestinal flora, the dosage form coated with this polymer would be effective for colon targeting of orally administered drugs. However, this poly-urathane based on m-xylene diisocynate (XDI), was soluble only in limited solvents and has been thought to be clinically inapplicable due to the trace amount of remaining solvent. Therefore Yamaoka et al. (125) synthesised a segmented polyurethane containing azo aromatic groups in the main chain by ratio of isophorone diisocyanate with a mixture of m, m'-di (hydroxymethyl) azobenzene, poly (ethylene glycol), and 1, 2-propanediol. This polyurethane was soluble in various solvents and showed a good coating and film-forming property. A solution-cast film of this polyurethane was found to be degraded in a culture of intestinal flora with the azo group reduction to hydroazo groups, not to amino groups. The film degradation, therefore, was attributed to the decreased cohesive energy in the hydroazo polymer compared with that in the original azo polymer. Then, the drug pellets containing water-soluble drugs were undercoated with (carboxymethyl) (ethyl) -cellulose and over coated with the azo polymer in order to examine the drug-releasing profiles in the culture of intestinal flora.
The release rate of drugs from these double-coating pellets was found to depend on the molecular weight and the composition of the polyurethane used as the overcoat as well as the hydrophilicity of the incorporated drugs. Since the polyurethane was glassy and its segment motion or conformational change was frozen, the structure change should be retarded even after partial reduction of the azo groups, resulting in the effective prevention of the drug leakage. Further, Chavan et al. (126) synthesised a urethane-based analogue containing an azo aromatic linkage in the backbone for use in colon-specific delivery systems by reacting toluene-2, 6-diisocyanate with a mixture of an aromatic azo diol, (bis-4-hydroxyphenyl)-4, 4'-diazobiphenyl, poly (ethylene glycol) and 1, 2-propanediol (propylene glycol). The compounds exhibited low molecular weight, lacked film-forming properties and crystallinity in the structure. An in vitro bacterial degradation test to demonstrate the susceptibility of azo bond to bacterial enzymes was performed using media inoculated with lactobacillus culture. The results indicated degradation of films by azoreductase. In vitro permeation of 5-ASA was studied in control and lactobacilli-treated films. The permeability of the lactobacilli treated films was significantly increased suggesting the potential of these compounds for application in colonic targeting. Lehmann and Drehher (127) used a suspension of natural polygalactomannans in polymethacrylate solutions to form degradable coating. The polygalactomannans form a swellable layer around the drug core, thus delaying the release of drug in the small intestine. They are destroyed enzymatically in the colon and consequently the drug is released.
In order to formulate inulin as a biodegradable coating material, it was incorporated as a suspension in Eudragit® RS films, since inulin itself has no film forming properties. Eudragit® RS, copolymer of acrylic acid esters with a low content of quaternary ammonium groups was chosen as film-former because it gives water-insoluble, pH-independent, low permeable films which are inert to endogenous digestive secretions and enzymes (128). The fall in pH was observed after incubation with human fecal medium for 24 h due to the formation of degradation products such as lactic acid, acetic acid and other volatile fatty acids. A significant increase in the permeability coefficient was detected after incubation within the control medium and on increasing the amount of inulin HP in the film the permeability coefficient showed a tendency to increase. Wakerly et al. (129) investigated the potential of pectin and ethyl cellulose (EC) combination as practical coatings for colon targeted delivery. Paracetamol cores were coated using aqueous dispersion consisted of pectin and EC. From the results of the study it was concluded that drug release was controlled by the reaction of EC to pectin in the film coat.
Bronsdsted and Hovgarrd (130) investigated the application of glutaraldehyde crosslinked dextran as a capsule material for colon-specific drug delivery. A reaction mixture containing dextran, magnesium chloride, glutaraldehyde and PEG 400 in water was applied onto moulding pins of nylon producing capsule caps and bodies. The capsule materials were characterised by measuring the mechanical strength in compression and equilibrium degree of swelling. Based on these results an optimal composition for the capsule material was selected. The dextran capsules were loaded with hydrocortisone and subsequently drug release was studied. The release was found to be about 10% during the initial 3 h in a buffer solution and over a period of 24 h the release was about 35%. However, when the dextran capsules were challenged with a dextranase solution, simulating the arrival of the drug delivery system to the colon, the capsules quickly broke and the drug was released as a dose dump. Bauer and Kesselhut. (131) proposed ester based on dextran with molecular weight of approximately 250 kD and degree of solubilization ranging from 0.11 to 0.4 as film forming and stable between pH 1.0 and 7.4. Tablet cores with theophylline were coated using conventional equipment with dispersion of 4% lauroyl dextran to theoretical polymer weight of 5, 10 and 15 mg/cm 2 (132). Dissolution of theophylline was carried out in a buffer of pH 5.5 for 4 h, after which dextranase was added to simulate the colonic environment. The release was inversely proportional to the amount of ester applied on the coatings. The entire degradation of coating was observed within 2 h after the addition of dextranase in the dissolution medium.
A novel oral delivery system for the treatment of IBD based on the microencapsulation of anti-inflammatory drugs, sulfasalazine and betamethasone using different biodegradable polymers, poly (-caprolactone), polylactic acid and poly(lactic-co-glycolic acid), was prepared either by the water-in-oil-in-water (w/o/w) or the solid-in-oil-in-water (s/o/w) solvent evaporation method (133). Microparticles were characterized for their size, morphology, encapsulation efficiency and drug release. In vitro release studies showed a controlled release of sulfasalazine and betamethasone from microparticles prepared by the s/o/w-method while a pronounced burst release of sulfasalazine was observed from microparticles prepared by the w/o/w method.
Embedding in matrices
The drug molecules are embedded in the polymer matrix. The polymers used for this technique should exhibit degradability in the colon for liberation of entrapped drug.
Embedding in biodegradable matrices and hydrogels
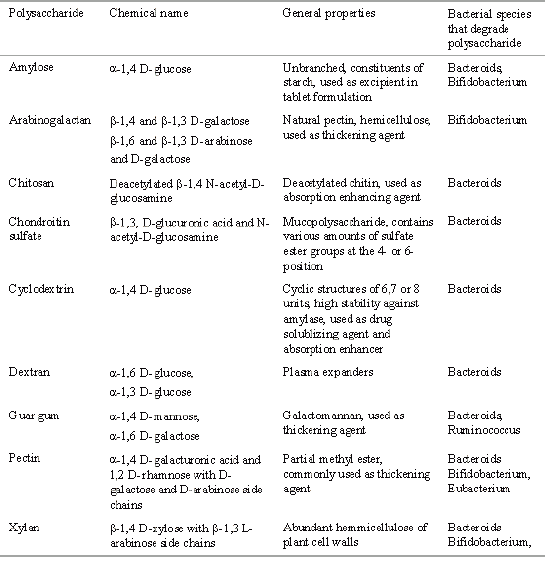
Polysaccharides, the polymer of monosaccharides retains their integrity because they are resistant to the digestive action of gastrointestinal enzymes. The matrices of polysaccharides are assumed to remain intact in the physiological environment of stomach and small intestine but once they reach in the colon, they are acted upon by the bacterial polysaccharidases and results in the degradation of the matrices. This family of natural polymers has an appeal to the area of drug delivery as it is comprised of polymers with a large number of derivatizable groups, a wide range of molecular weights, varying chemical compositions, and for the most part, a low toxicity and biodegradability, yet a high stability. The most favorable property of these materials is that they are already approved as pharmaceutical excipients. A large number of polysaccharides such as amylose, guar gum, pectin, chitosan, inulin, cyclodextrins, chondroitin sulphate, dextrans and locust bean gum have been investigated for their use in colon targeted drug delivery systems. The most important fact in the development of polysaccharide derivatives for colon targeted drug delivery is the selection of a suitable biodegradable polysaccharide. As these polysaccharides are usually soluble in water, they must be made water insoluble by crosslinking or hydrophobic derivatisation. Very important is an optimal proportional of the hydrophobic and hydrophilic parts respectively and the number of free hydroxy groups in the polymeric molecule. General properties of polysaccharides used in colon targeted drug delivery are shown in Table 4.
Table 4: Characteristics of various biodegradable polysaccharides for colon targeted drug delivery.
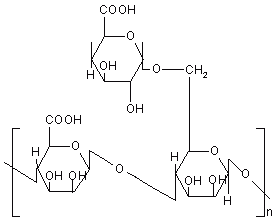
Pectin is a polysaccharide consists of a-1, 4 D-galacturonic acid and 1, 2 D-rhamnose with D-galactose and D-arabinose side chains (Figure 8). A novel colonic drug delivery system based on the polysaccharide pectin has been investigated. In vitro experiments demonstrated that high methoxy pectin, when applied as a compression coat, proved capable of protecting a core tablet during conditions stimulating gastrointestinal environment and was susceptible to enzymatic attack.
Figure 8: Chemical structure of pectin.
In vivo gamma scintigraphic studies confirmed the in vitro findings. In all the volunteers, the pectin-coated tablets disintegrated in the colon indicating that site-specificity had been achieved and illustrating the potential of a colonic drug delivery system utilizing pectin. However, in the in vivo conditions, a coat of considerable thickness was required to protect the drug core. This necessitates the development of such derivatives of pectin, which were less water-soluble but were having the capability to be degraded by the colonic microflora. Calcium pectinate, the insoluble salt of pectin was used for colon targeted drug delivery of indomethacin by Rubeinstein et al. (134). The use of calcium pectinate as a carrier was based on the assumption that, like pectin, it can be decomposed by specific pectinolytic enzymes in the colon but retains its integrity in the physiological environment of small bowel. In spite of reduction of water solubility, calcium pectinate compressed with indomethacin into tablets was shown to degrade by enzymes of Aspergillus and colonic bacteria Bacteroides ovatus as reflected from the in vivo performance studies (135). Ashford et al. (136) prepared matrix tablets of a model compound sodium fluorescein and assessed in vitro , the potential of several pectin formulations as colon targeted drug delivery systems. Various variables like pectin, the presence of calcium and the solubility of the calcium salt influenced the release of drug. It was observed that either a high methoxy pectin formulation or low methoxy pectin with a carefully controlled amount of calcium maximized the colonic specificity by providing optimal protection of drug during its transit to the colon and a high susceptibility to enzymatic degradation. Combination of the pectin along with either chitosan or HPMC has been successfully used for the formulation of colon targeted drug delivery systems.
Combination of pectin and ethyl cellulose film was used for colonic delivery of paracetamol (137, 138). Aqueous dispersions of pectin and ethylcellulose were used to film coat paracetamol tablet cores and drug release mechanisms were assessed using flow through dissolution testing in the presence and absence of enzymes. Drug release from the coated systems was complex and depended on the nature and characteristics of the mixed film as well as the composition of the dissolution medium. Drug release profiles were compatible with a mechanism involving the formation of channels in the film caused by pectin dissolution. Channel formation was accelerated in most of the cases by the presence of pectinolytic enzymes showing that the pectin in the mixed film was susceptible to enzymic attack. The mechanical and permeability properties of mixed ethylcellulose/pectin films cast from dibutyl sebacate plasticised aqueous dispersions of Aquacoat® and Pectin USP has been investigated. The films were subjected to tensile testing, elongation at break and elastic modulus. Increasing concentrations of pectin imparted increasing brittleness and decreasing toughness to the films. Despite the inclusion of increasing quantities of the hydrophilic pectin into the films, the permeability to moisture remained essentially the same. The results imply that there is a limit to the amount of pectin that can be included in the coating material to still produce a satisfactory film, but the protective nature of the ethylcellulose to moisture is not compromised. Fernandez-Hervás and Fell (139) used pectin and chitosan mixtures as coatings for colon-specific drug delivery of indomethacin and paracetamol which were used as model drugs to represent poorly soluble and soluble compounds, respectively. Pectin alone was able to protect the cores from premature release, but only when a substantially thick coat was present. Pectin/chitosan mixtures achieved better protection at a lower coat weight. The use of pectinolytic enzymes to simulate breakdown in the colon showed that the pectin/chitosan mixture was susceptible to enzymic breakdown and allowed drug release to occur. McLeod et al. (140) carried out a study to assess the potential of pectin:chitosan films for colonic drug delivery. Turkoglu and Ugurlu (141) reported pectin-HPMC compression coated core tablets of 5-ASA for colonic delivery. Drug dissolution/system erosion/degradation studies were carried out in pH 1.2 and 6.8 buffers using a pectinolytic enzyme. The system was designed based on the gastrointestinal transit time concept, under the assumption of colon arrival times of 6 h. It was found that pectin alone was not sufficient to protect the core tablets and HPMC addition was required to control the solubility of pectin. The optimum HPMC concentration was 20% and such system protected the cores up to 6 h that corresponded to 25-35% erosion and after that under the influence of pectinase the system degraded faster and delivered 5-ASA to the colon. Combination of pectin (pectin HM or calcium pectinate) and insoluble polymer aqueous dispersions for specific delivery of drugs to the colon has reported by Samde et al. (142, 143). Theophylline pellets were coated with cellulosic (Aquacoat ECD 30, Surelease clear) or acrylic (Eudragit® NE30D, RS30D) polymer aqueous dispersions, containing 10% (related to the insoluble polymer content) of pectin HM or calcium pectinate, using a Uni-Glatt fluidized-bed coating apparatus. The results obtained have been examined with regard to the validity of the approach based on the combination of pectin and the insoluble polymer aqueous dispersions intended for specific-delivery of drugs to the colon.
Amidated pectins are low-methoxy pectins in which some of the carboxylic acid groups are amidated. They are more tolerant of pH variations and calcium levels than conventional pectins, which could make them useful in colonic delivery systems. Gelation of droplets in presence of calcium may provide a valuable approach to the formation of a multiparticulate system for colonic delivery. The properties of such amidated pectin beads may be altered by the formation of a polyelectrolyte complex membrane around the bead using cationic polymers such as chitosan (144). Despite the presence of chitosan in the amidated pectin beads, it retained its ability to degradation by pectinolytic enzymes and significantly reduced the release of sulfamethoxazole and indomethacin in simulated gastric and intestinal fluid as compared with non-amidated beads. Similarly Sriamornsak et al. (145) prepared calcium pectinate gel (CPG) beads of indomethacin, by dispersing the drug in a solution of pectin and then dropping the dispersion into calcium chloride solution. The droplets instantaneously converted into gelled spheres by ionotropic gelation. The effect of several factors such as pectin type, the presence of a hardening agent and the drug loading were investigated on the percentage of drug entrapped, size distribution and drug release from the CPG beads. El-Gibaly et al. (146) proposed oral delayed-release system based on Zinc-pectinate gel microparticles as an alternative carrier to calcium pectinate beads for colonic drug delivery. The system, which consists of ketoprofen-loaded Zinc-pectinate gel (ZPG) microparticles together with pectin/dextran mixtures in a tablet form, has been investigated, in vitro , using conditions chosen to simulate the pH and times likely to be encountered during transit to the colon. In order to find the suitable ZPG microparticles, the formulations were prepared by utilizing 2(3) factorial design and the effect of various formulation factors on the release and surface characteristics of the microparticles was studied. The results obtained implied that the release of ketoprofen from ZPG microparticles was greatly extended with the pectinate microparticles, which were prepared with 2.5 or 3% w/v pectin, 2.75% w/v zinc acetate and 2.5% w/v drug. Additionally, the analysis of variance results showed that the release of ketoprofen in simulated intestinal fluid (pH 7.4) was strongly affected by crosslinking agent concentration and initial drug amount, but not particularly affected by the amount of pectin added. The investigated drug concentration factor has significantly increased the drug entrapment efficiency. The optimum colonic drug delivery ZPG/tablet system provided the expected delayed-release sigmoidal patterns with a lag-time of 4.125-4.85 h and t50% (the time for 50% of the drug to be released) at 7.45-8.70 h, depending on pectin/dextran ratio employed. The results also demonstrated that the untableted ZPG microparticles exhibited drug release profiles that were able to retard the release of ketoprofen in simulated intestinal fluid (pH 7.4) to be 5.28-37.82 times (depending on formulation parameters) lower than the conventional calcium pectinate beads.
Hydrogels are usually formed by the covalent crosslinking of linear hydrophilic polymers to form a network of material capable of absorbing water, yet still remaining insoluble (147). Heterogenous polymer mixtures may also be used to form hydrogels without the need for covalent crosslinking (148). Various hydrogels based on the azo polymeric networks have been developed for site-specific delivery of drugs to the colon (Table 5).
Table 5: Various hydrogels for colon targeted drug delivery
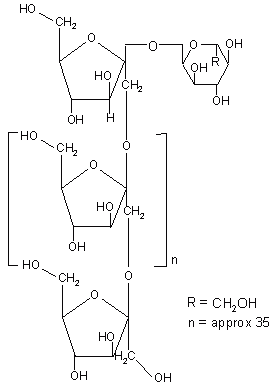
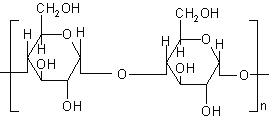
Inulin is a naturally occurring polysaccharide found in many plants. It consists of β 2-1 linked D-fructose molecules having a glucosyl unit at the reducing end (Figure 9). Various inulin and dextran hydrogels have been developed that serve as potential carrier for introduction of drugs into the colon (Table 5).
Figure 9: Chemical structure of inulin.
Poly vinyl alcohol, of different molecular weights were cross-linked with succinyl, adipoyl, or sebacoyl chloride to obtain hydrogel-forming polymers and their suitability as colon-specific drug delivery systems was determined. The results indicated the ability of the cross-linked polymers to slow the release of the drugs analyzed with respect to the pure drug dissolution at each pH. The lengthening of the cross-linker acyl chain was noted to decrease drug release further (162). A new series of water insoluble acrylic polymers based on cellobiose-derived monomers for colon targeting was reported (163). In addition, water-soluble acrylic polymers such as Carbopol 974P were also evaluated for the controlled intestinal delivery of mesalamine (164). Cen et al. (165) prepared pH sensitive poly (acrylamide/maleic acid) hydrogels for controlled release of terbinafine hydrochloride. In vitro drug release studies in different buffer solutions showed that the basic parameters affecting the drug release behaviour of hydrogel were the pH of the solution and MA content of hydrogel.
Guar gum is a polysaccharide derived from the seeds of Cyamopsis tetragonolobus and many reports in the literature has proved its efficacy for colonic drug delivery. It consists of linear chains of (1→ 4)-b-D-manopyranosyl units with α-D-galactopyranosyl units attached by (1→ 6) linkages (Figure 10) (166).
Figure 10: Chemical structure of guar gum.
Guar gum is hydrophilic in nature and swells in cold water forming viscous colloidal dispersions or sols (167). This gelling property retards release of the drug from the dosage form as well as it is susceptible to degradation in the colonic environment. Homogenized and diluted feces from human source were incubated with the guar gum to investigate the degradation of polysaccharide by intestinal microflora. It produced a rapid decrease in viscosity and fall in pH while no such results were observed when it was incubated with autoclaved fecal homogenates (168). Guar gum was crosslinked with increasing amounts of trisodium trimetaphosphate to reduce its swelling properties for use as a vehicle in oral delivery formulations. As a result of the crosslinking procedure guar gum lost its non-ionic nature and became negatively charged. This was demonstrated by methylene blue adsorption studies and swelling studies in sodium chloride solutions with increasing concentrations in which the hydrogels' network collapsed (169). Crosslinked guar gum products were analysed to check the efficacy as colon-specific drug carrier and it was found that the product which was crosslinked with 0.1 equivalent of trisodium trimetaphosphate was able to prevent the release of 80% of its hydrocortisone load for at least 6 h in PBS (pH 6.4). When a mixture of a -galactosidase and b-mannanase was added to the buffer solution, an enhanced release was observed. In vivo degradation studies in the rat caecum showed that despite the chemical modification of guar gum, it retained its enzyme-degrading properties in a crosslinker concentration dependent manner (170). A novel tablet formulation for oral administration using guar gum as the carrier and indomethacin as a model drug has been investigated for colon targeted drug delivery using in vitro methods. Drug release studies under conditions simulating the gastrointestinal transit have shown that guar gum protects the drug from being released completely in the physiological environment of stomach and small intestine. Studies in pH 6.8 PBS containing rat caecal contents have demonstrated the susceptibility of guar gum to the colonic bacterial enzyme action with consequent drug release (171). Gilko Kabir et al. (172) in a study reduced the swelling properties of guar gum by crosslinking it with increasing amount of glutaraldehyde under acidic conditions and evaluated the degradation properties of modified guar gum. Reduction in the enormous swelling by crosslinking resulted in biodegradable hydrogel formation, which was able to retain poorly water-soluble drug. Krishnaiah et al. (173) studied the influence of metronidazole and tinidazole on the usefulness of guar gum, a colon-specific drug carrier based on the metabolic activity of colonic bacteria, using matrix tablets of albendazole (containing 20% of guar gum) as a model formulation. The guar gum matrix tablets of albendazole were found degraded by colonic bacteria of rat caecal contents and released about 44% of albendazole in simulated colonic fluids (control study) at the end of 24 h indicating the susceptibility of the guar gum formulations to the rat caecal contents.
Compression coated tablets (174) of 5-ASA and matrix tablets of mebendazole have been prepared using guar gum as a carrier. Matrix tablets containing various proportions of guar gum were prepared by wet granulation technique using starch paste as a binder. The tablets were evaluated for drug content uniformity, and were subjected to in vitro drug release studies. The results of the study revealed that matrix tablets containing either 20% or 30% of guar gum are most likely to provide targeting of mebendazole for local action in the colon. A novel colon-specific drug delivery system based on guar gum matrix tablets was evaluated by conducting gamma scintigraphy studies using technitium-99m-DTPA as a tracer, in six healthy male human volunteers. Scintigraphs taken at regular intervals showed that some amount of tracer present on the surface of the tablets was released in stomach and small intestine and the bulk of the tracer present in the tablet mass delivered to the colon (175). Amylose is a polysaccharide obtained from plant extracts and a component of starch. It consists of D-glucopyranose residues linked by a -(1-4) bonds. It is a poly (1-4-a-D-glucopyranose) (Figure 11).
Figure 11: Chemical structure of amylose.
Colon-specific drug delivery may be possible by the application of dried amylose films to pharmaceutical formulations. Amylose, one of the major fractions of starch, possesses the ability to form films through gelation, when prepared under appropriate conditions. The microstructure of the film is potentially resistant to the action of pancreatic α-amylase but is digested by amylases of the colonic microflora. However, under simulated gastrointestinal conditions, coatings made solely of amylose will become porous and allow drug release. Incorporation of insoluble polymers into the amylose film, to control amylose swelling, provides a solution to this problem. A range of cellulose and acrylate based copolymers were assessed, of which a commercially available ethylcellulose (Ethocel) was found to control the swelling most effectively. The in vitro dissolution of various coated pellets under simulated gastric and small intestinal conditions, using commercially available pepsin and pancreatin was determined and demonstrated the resistance of the amylose-Ethocel coat (1:4) to such conditions over a period of 12 h (176). Glucose as a model drug was incorporated into pellets that were prepared by extrusion and spheronization to assess the colonic drug delivery capability. The behaviour of different glucose containing pellets coated with an amylose-Ethocel mixture was investigated in vitro and formulation was found to be gastric and small intestine resistant. In vitro fermentation studies revealed that the formulation was susceptible to bacterial enzymatic attack (177). Epichlorhydrin treated crosslinked amylose was introduced as a matrix for controlled release of theophylline (178). A mixture of amylose and Ethocel (1:4) has been developed and [ 13 C] glucose used as surrogate for drug delivery (179).
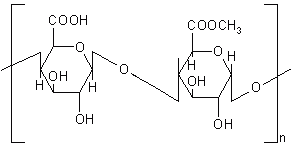
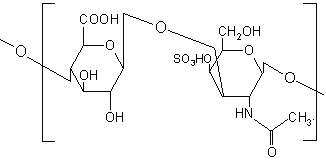
Chondroitin sulphate is a mucopolysaccharide, which consists of D-glucuronic acid linked to N-acetyl-D-galactosamide (Figure 12). It is degraded by the anaerobic bacteria of the large intestine mainly by Bacteroids thetaiotaoimicron and B. ovatus (180). The high water solubility of chondroitin sulphate put hurdles in the formulation of colon targeted drug delivery systems and hence crosslinking has been reported in the literature to alleviate this problem. Chondroitin sulphate was cross-linked with 1, 12 diaminododecane using dicyclohexylcarbodiimide as a catalyst and formulated in a matrix with indomethacin as a drug marker. The indomethacin release kinetics from the various formulations was analysed in PBS with and without rat caecal content at 37°C under carbon dioxide atmosphere and it was concluded that release of indomethacin was dependent upon the biodegradation action of the caecal content (181, 182). Sintov et al. (183) cross-linked chondroitin sulphate with 1, 12-diaminododecane to give a series of cross-linked products with reduced water solubility. Indomethacin tablets were prepared using two types of cross-linked polymers; very low water soluble and relatively high water soluble and analysed for their water uptake and drug release characteristics.
Figure 12: Chemical structure of chondroitin sulphate.
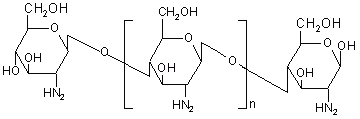
Chitosan is a high molecular weight polycationic polysaccharide derived from naturally occurring chitin by alkaline deacetylation. Chemically, it is a poly (N-glucosamine) (Figure 13). Chitosan has favourable biological properties such as nontoxicity, biocompatibility and biodegradability. Similar to other polysaccharides it also undergoes degradation by the action of colonic microflora and hence poses its candidature for colon targeted drug delivery. Tozaki et al. (184, 185) developed colon-specific insulin delivery with chitosan capsules. In vitro drug release experiments from chitosan capsules containing 5(6)-carboxyfluorescein (CF) were carried out by rotating basket method with slight modifications. The intestinal absorption of insulin was evaluated by measuring the plasma insulin levels and its hypoglycaemic effects after oral administration of the chitosan capsules containing insulin and additives. Little release of CF from the capsules was observed in an artificial gastric juice (pH 1), or in an artificial intestinal juice (pH 7). However, the release of CF was markedly increased in the presence of rat caecal contents.
Figure 13: Chemical structure of chitosan.
The naturally occurring polymer chitosan was reacted separately with succinic and phthalic anhydrides. The resulting semisynthetic polymers were assessed as potential matrices for colon-specific, orally administered drug delivery using sodium diclofenac as the dispersed model drug. The prepared matrices were incorporated into tablets, evaluated in vitro which resisted dissolution under acidic conditions. On the other hand, improved drug release profiles were observed under basic conditions that suggest the suitability of the prepared matrices in colon-specific, orally administered drug delivery system (186). Therapeutic effect of R-68070, a new thromboxane synthetase inhibitor on 2,4,6 trinitrobenzene sulfonic acid sodium salt induced ulcerative colitis was studied using chitosan capsules to achieve its colon-specific drug delivery in rats (187). Tozaki et al. (188) evaluated colon-specific insulin delivery using chitosan capsules. It was found that these were stable in the stomach and small intestine but degraded by micro-organism in rat caecal contents upon entering into the colon proving their utility as carriers for colon targeted drug delivery of peptide and nonpeptide drugs. A pH-sensitive drug delivery carrier has also been reported for chitosan-based hydrogels (189, 190). A multiparticulate system of chitosan hydrogel beads has been investigated for colon-specific drug delivery of macromolecules using fluorescein isothiocanate-labeled bovine serum albumin as a model protein. The hydrogel bead was formed by polyelectrolyte complexation of chitosan with its counter ion, tripolyphosphare. The protein release experiments were carried out in vitro under different conditions to simulate the pH and times likely to be encountered during intestinal transit to the colon. The results shown that the hydrogel beads were degraded by rat cecal and colonic enzymes, resulting in a marked acceleration in the release of protein (191). Chitosan dispersed drug delivery system which was composed of active ingredient reservoir and the outer drug release regulating layer dispersing chitosan powder in hydrophobic polymer, was newly developed for colon-specific drug delivery (192). Different chitosan salts were prepared by dissolving in aqueous solutions containing aspartic, glutamic, hydrochloric, lactic and citric acid. The in vitro evaluation was carried out to study the influence of acid type on the release behavior of incorporated diclofenac sodium from the physical mixture during gastrointestinal transit. The physical mixture of chitosan salts with diclofenac sodium provided slower drug release than the pure drug both in acidic and alkaline pH and in addition presence of b-glucosidase at pH 7.0 enhanced the release rate (193).
Embedding in pH-sensitive matrices
Extrusion-spheronization and pelletization have been used for the preparation of pH-sensitive matrix pellets for colon targeted drug delivery (194). The authors studied the effects of three independent variables (amounts of Eudragit® S, citric acid and spheronizing time) on pellet size, shape (roundness and aspect ratio), and drug release was studied with central composite design. Nykanen et al. (195) used ibuprofen as model drug and Eudragit® S and Aqoat AS-HF as enteric polymers for developing site-specific systems for release of a drug in the lower part of the small intestine or in the colon. The target of this study was to investigate whether drug release rate from enteric matrix granules could be influenced by using organic acids as excipients. It was concluded that although inclusion of an organic acid in a formulation retarded in vitro release of the model drug, no corresponding effect was evident in case of in vivo studies.
Timed release systems
This approach is based on the principle of delaying the release of the drug until it enters into the colon. Although gastric emptying tends to be highly variable, small intestinal transit time is relatively constant or little bit variation can be observed. The strategy in designing timed-released systems is to resist the acidic environment of the stomach and to undergo a lag time of predetermined span of time, after which release of drug take place. The lag time in this case is the time requires to transit from the mouth to colon. The first formulation introduced based on this principle was Pulsincap® (196). It is similar in appearance to hard gelatin capsule; the main body is made water insoluble (exposing the body to formaldehyde vapour which may be produced by the addition of trioxymethylene tablets or potassium permanganate to formalin or any other method). The contents are contained within a body by a hydrogel plug, which is covered by a water-soluble cap. The whole unit is coated with an enteric polymer to avoid the problem of variable gastric emptying. When the capsule enters the small intestine the enteric coating dissolves and the hydrogels plug starts to swell, the amount of hydrogel is such adjusted that it pops out only after the stipulated period of time to the release the contents. The viability of such a system in human volunteers has been confirmed on the basis of evaluation studies (197, 198). A multiple coated oral dosage form consisting of core coated with three polymeric layers has developed (199, 200). Gazzaniga et al. described a novel oral time based drug release system for colon-specific delivery. The system designed to exploit the relatively constant small intestinal transit time of dosage forms consists of drug-containing cores coated with three polymeric layers. The outer layer dissolves at pH > 5, then the intermediate swellable layer, made of an enteric material. The system provides the expected delayed release pattern, as also indicated by the preliminary in vivo studies on rats. Several other drug delivery systems have developed that rely upon the relatively constant transit time of small intestine (201-205). A novel delivery system was developed for delivering drugs to the colon by selecting polymethacrylates with appropriate pH dissolution characteristics for the distal end of the small intestine. Pellets were prepared by powder layering of 5-ASA on nonpareils (0.5-0.6 mm) in a conventional coating pan. Drug-layered pellets were coated with an inner layer of a combination of two pH-independent polymers Eudragit® RL and RS (2:8), and an outer layer of a pH-dependent polymer, Eudragit® FS. Scanning electron micrograph pictures of the coated pellets showed the uniformity of both the coatings. The release profile of 5-ASA was studied in three phosphate buffers after a simulated gastric pre-soak for 2 h in pH 1.2 media. There was no drug release for 12 h at pH 6.5. There was a sustained release of 5-ASA for over 12 h both at pH 7.0 and 7.5 after a lag time at pH 7.0 and no lag time at pH 7.5. The release rate was faster at pH 7.5 than at pH 7.0. The delivery system demonstrated its potential for colonic delivery by resisting drug release until pH 6.5 and the combination of Eudragit® RL and RS proved successful for the sustained delivery of 5-ASA at the expected pH of the colon (201). As a new oral drug delivery system for colon targeting, enteric coated timed-release press-coated tablets (ETP tablets) were developed by coating enteric polymer on timed-release press-coated tablets composed of an outer shell of hydroxypropylcellulose and core tablet containing diltiazem hydrochloride as a model drug. The results of the in vitro dissolution tests in JP 1st fluid (pH 1.2) and JP 2nd fluid (pH 6.8) indicated that these tablets showed both acid resistance and timed-release characteristics. To clarify whether ETP tablets could have been of use in the gastrointestinal tract, ETP tablets with a layer of phenylpropanolamine hydrochloride (PPA) (a marker of gastric emptying) between the enteric coating layer and outer shell were prepared, and were administered to beagle dogs. The gastric emptying time and lag time after gastric emptying was evaluated by determining the times at which PPA and diltiazem hydrochloride first appeared in the plasma (202). To develop a new colon targeting formulation, which can suppress drug release completely during 12 h in the stomach and release the drug rapidly after a lag time of 3±1 h in the small intestine, the use of press-coated tablets with hydroxypropylmethylcellulose acetate succinate (HPMCAS) in the outer shell was investigated. The release of diltiazem hydrochloride as a model drug contained in the core tablets in the 1st fluid (pH 1.2) was suppressed by preparing with higher compression force, but the lag time in the 2nd fluid (pH 6.8) could not exceed 1.5 h. Therefore, to improve the dissolution characteristics, the effects of addition of various hydrophobic additives to HPMCAS were examined. All of the additives examined suppressed the release rate in the 1st fluid, and prolonged the lag time in the 2nd fluid compared to HPMCAS alone. However, none of the additives examined fulfilled all of the desired criteria, magnesium stearate and calcium stearate showed interesting effects; the former suppressed drug release completely in 1st fluid, while the latter markedly prolonged the lag time in 2nd fluid. To integrate the merits of each additive, press-coated tablets with a powder mixture of HPMCAS, magnesium stearate and calcium stearate in the outer shell were prepared and in vitro tests were performed.
In another method, an organic acid (succinic acid) was filled into the body of a hard gelatine capsule as a pH-adjusting agent together with the drug substance. The joint of the capsule was sealed using an ethanolic solution of ethylcellulose. The capsule was first coated with an acid soluble polymer (Eudragit® E), then with a hydrophilic polymer HPMC and finally enterically coated with Eudragit® L. After ingestion of the capsule, the outermost enteric layer of the coating prevents drug release in the stomach. Enteric layer and hydrophilic layers dissolve quickly after gastric emptying and water starts entering the capsule. When the environmental pH inside the capsule decreases by the dissolution of organic acid, the acid soluble layer dissolves and the enclosed drug is quickly released. Therefore, the onset time of drug releases in the intestine can be controlled by the thickness of acid soluble layer (203).
A delivery system called the Time Clock® has been exploited to release the drug in the colon (206). It is composed of a solid dosage form coated with a hydrophobic surfactant layer to which a water-soluble polymer is added to improve adhesion to the core. The outer layer redisperses in the aqueous environment in a time proportional to the thickness of the film and the core is then available for dispersion. In a study with human volunteers, it was shown that the lag time was independent of gastric residence time and hydrophobic film redispersing did not appear to be influenced by the presence of intestinal digestive enzymes or by mechanical action of the stomach. A capsule consisting of EC was prepared and evaluated for site-specific drug delivery to the colon (207). It is composed of a low substituted hydroxy propyl cellulose drug container, a capsule body and a capsule made of EC. Water penetrates through the micropores presents at the bottom of capsule and the swelling of polymer forces the EC cap to disintegrate, thereby releasing the drug.
Pressure controlled drug delivery systems have developed to target the drugs to the colon (208-211). Pressure-controlled colon delivery capsule (PCDC) made of EC was prepared by coating the inner surface of gelatin capsule with water-insoluble polymer, EC. By adjusting the coating thickness of EC membrane to be approximately 40 microns, colon delivery of drug were obtained both in beagle dogs and human volunteers. PCDC containing 5-ASA was prepared and was administered orally to beagle dogs. After administration, 5-ASA appeared into the systemic circulation at 3-5 h that corresponds to the colon arrival time confirmed with sulfasalazine (208). The relationship between in vitro drug release characteristics from colon delivery systems and in vivo drug absorption was investigated using three kinds of delayed-release systems. 5-ASA, tegafur and carbamazepine were selected as model drugs. Pressure-controlled colon delivery capsules for liquid preparations, time-controlled colon delivery capsules for liquid and solid preparations and Eudragit® S coated tablets for solid preparations were used in this study. At first, in vitro dissolution tests for all preparations were performed. Drug release from solid preparations was delayed compared to that from liquid preparations with all three drugs. From the findings of the study it was concluded that drug release from colon delivery systems and drug dissolution in the colonic lumen are very important factors for the systemic availability of drugs from the colon delivery systems (209). The delivery ability of a PCDC containing caffeine as a test drug was evaluated after oral administration to healthy male human volunteers. The driving force causing PCDC disintegration in the intestinal tract is the physiological luminal pressure, which results from peristalsis. Three kinds of PCDCs having different thickness of a water-insoluble polymer membrane were prepared by coating the inner surface of the gelatin capsules with EC. The mean thicknesses were 40 ± 1 mm for type 1, 44 ± 1 mm for type 2 and 50 ± 1 mm for type 3 PCDC, respectively. Caffeine was dissolved with a suppository base (PEGs 400 and 1000) and the capsules were filled in doses of 15, 45 or 75 mg. After blank saliva samples were obtained, test preparations were orally administered to the volunteers and saliva samples were collected for 1 min intervals hourly from 1 to 10 h in the fasted state study, and from 1 to 20 h and at 25 h in the fed state study. Caffeine concentrations in the saliva samples were analysed by HPLC. The maximum salivary caffeine excretion rate increased as the oral caffeine dose increased (210).
Evaluation of an oral system (Chronotopic) designed to achieve time and/or site-specific release has been reported by Sangalli et al. (212). The system consists of a drug-containing core, coated by a hydrophilic swellable polymer, which is responsible for a lag phase in the onset of release. In addition, through the application of an outer gastro resistant film, the variability in gastric emptying time can be overcome and a colon-specific release can be sought relying on the relative reproducibility of small intestinal transit time. Cores containing antipyrine as the model drug were prepared by tableting and both the retarding and enteric coatings were applied in fluid bed. The in vitro release curves presented a lag phase preceding drug release and the in vivo pharmacokinetic data showed a lag time prior to the detection of model drug in saliva. Both in vitro and in vivo lag times correlate well with the applied amount of the hydrophilic retarding polymer. The scintigraphic study pointed out that the break-up of the units occurred in the colon. The results showed the capability of the system in delaying drug release for a programmable period of time and the possibility of exploiting such delay to attain colon targeted delivery according to a time-dependent approach.
Redox-sensitive polymers
Analogues to azo bond cleavage by intestinal enzymes, novel polymers that hydrolyzed nonenzymatically by enzymatically generated flavins are being developed for colon targeting. Biodegradation of azo polymers has been extensively studied in the literature (213, 214). It is suggested that both an intracellular enzymatic component and extracellular reduction exist. Under anaerobic conditions, bacterial azo reduction by enzymatically generated reduced flavins where the initial substrate thought to be involved in cellular electron transport requires the presence of NADPH as its electron source. As NADPH is oxidized, the electron mediator (reduced flavins) acts as an electron shuttle from the NADPH dependent flavoprotein to the azo compound. Molecular modeling of low molecular weight azo compounds revealed that reduction of the azo bond to the hydroazo intermediate requires a low electron density within the azo region, and thus substitution of electron-withdrawing groups will favor this reaction. Redox potential is an expression of the total metabolic and bacterial activity in the colon and it is believed to be insensitive to dietary changes. The mean redox potential in proximal small bowl is - 67 ± 90 mv, in the distal small bowl is -196 ± 97 mv and in the colon is -145 ± 72 mv. Thus, microflora-induced changes in the redox potential can be used as a highly selective mechanism for targeting to the colon. Bragger et al. (215) carried out investigations into the azo reducing activity, which could enlighten some factors affecting the bacterial reduction (cleavage) of azo compounds. A common colonic bacterium, Bacteroides fragilis was used as test organism and the reduction of azo dyes amaranth, Orange II, tartrazine and a model azo compound, 4, 4'-dihydroxyazobenzene were studied. It was found that the azo compounds were reduced at different rates and the rate of reduction could be correlated with the redox potential of the azo compounds. 4,4'-Dihydroxyazobenzene (E1/2 -470 mV) was reduced at the fastest rate of 0.75 mol l -1 h -1, amaranth (E1/2 -568 mV) at 0.30 mol l-1 h -1, Orange II (E1/2 -648 mV) at 0.2 mol l -1 h -1 and tartrazine (E1/2 -700 mV) at 0.08 mol l-1 h-1. Similar observations were made with another colonic bacterium Eubacterium limosum .
Disulphide compounds can also undergo degradation due to the influence of redox potential in the colon (216). Noncrosslinked redox-sensitive polymers containing an azo and/or a disulfide linkage in the backbone have been synthesised (217). Radiological studies in dogs have investigated the in vitro behaviour of new polyurethane systems containing azo bonds (218, 219).
Bioadhesive systems
Oral administration of some drugs requires high local concentration in the large intestine for optimum therapeutic effects. Dissolution of dosage form and simultaneous absorption from upper GIT lead to low intracolonic drug concentration as well as absorption of drugs result in the generation of side effects. Bioadhesion is a process by which a dosage form remains in contact with particular organ for an augmented period of time. This longer residence time of drug would have high local concentration or improved absorption characteristics in case of poorly absorbable drugs. This strategy can be applied for the formulation of colonic drug delivery systems. Various polymers including polycarbophils, polyurethanes and polyethylene oxide-polypropyline oxide copolymers have been investigated as materials for bioadhesive systems (220, 221). Bioadhesion has been proposed as a means of improving the performance and extending the mean residence time of colonic drug delivery systems (222, 223). In vitro bioadhesion has been confirmed from many studies and few reports are available in the literature regarding the in vivo bioadhesion studies (224, 225). Kakoulides et al. (226, 227) synthesized Azo-networks based on an acrylic backbone crosslinked with DVAB (Figure 14). The chemical structure of the synthesised series of copolymers was examined by infrared spectroscopy and nuclear magnetic resonance data. The thermal properties of the materials were assessed using a combination of thermal analysis techniques and their swelling behaviour was evaluated at physiologically relevant buffers designed to mimic the gastrointestinal environment. These networks were subjected to in vitro degradation and mucoadhesion (before and after degradation) testing in order to model their performance in the gastrointestinal tract. Advanced surface characterisation techniques (SEM, AFM, FTIR microscopy) were used to examine the network morphology prior to, and after degradation. These studies indicate that there is an optimum crosslinking density to allow non-adhesive particles to reach the colon. Within the colonic environment, the azo network degrades to produce a structure capable of developing mucoadhesive interactions with the colonic mucosa.
Figure 14: Mucoadhesive azo crosslinked acrylic acid and predicted colonic degradation behaviour.
Amino acids and polymers have used as drug carriers for colon targeted delivery of 5-ASA (228). Rihova used bioadhesive polymers such as HPMA copolymers that are used to mimic bioadhesive process occurring in the guinea pig GIT which are based on the presence of lectin like structures on enterocytes and in the mucus gel layer (229). A water-soluble polymer containing salicylate residues azo-linked at the 5-position to the polymer backbone was synthesized for the treatment of IBD (230).
The design of targetable water-soluble polymeric drug carriers based on N- (2-hydroxy propyl) methacrylamide (HPMA) copolymers was described by Kopecek (231). A new concept of oral drug delivery was proposed based on a combination of site-specific delivery of 5-ASA to the colon with bioadhesive properties of the carrier. HPMA copolymers containing saccharide units complementary to mucosal lectins of the GIT are used as carriers. They also contain side chains terminated in salicylic acid bound via an azo bond. Cleavage experiments were carried out using an isolated strain of bacteria commonly found in the colon. When inoculated with Streptococcum faecium in vitro 5-ASA was released. Body distribution in guinea pigs after oral administration has shown that HPMA copolymers containing fucosylamine associate with the colon. HPMA copolymers were evaluated as colon-specific drug carriers. Their design was based on the concept of site-specific binding of carbohydrate moieties complementary to colonic mucosal lectins and on the concept of site-specific drug (5-ASA) release by the microbial azoreductase activity present in the colon. A new 5-aminosalicylic acid-containing monomer was synthesised and incorporated into the copolymer together with the fucosylamine bioadhesive moiety containing comonomer by radical copolymerisation (Figure 15).
Figure 15: Synthesis of N-(2-hydroxypropyl) methacrylamide copolymers by radical copolymerization.
The in vitro release rate of 5-ASA from HPMA copolymers by azoreductase activity in guinea pig caecum was approximately 2.5 times lower than from a low molecular weight analogue. The azoreductase activities in caecum contents of guinea pig, rat and rabbit as well as in human faeces were determined. The relative activities for rat:guinea pig:human:rabbit were100:65:50:28. Both in vitro and in vivo HPMA copolymer-containing side chains terminated in fucosylamine showed a higher adherence to guinea pig colon while compared to HPMA copolymer without fucosylamine moieties. The incorporation of 5-ASA containing aromatic side-chains into HPMA copolymer further increased their adherence probably by combination of non-specific hydrophobic binding with specific recognition (232).
Coating with microparticles
Many of the protozoans especially Entamoeba histolytica remains confined in the large intestine, which necessitates high intracolonic drug concentration (233). This goal is not fulfilling with the current available therapy, as they are based on the principle of releasing drugs into upper GIT that is systemically absorbed and generates side effects. Mirelman et al. (234) prepared and evaluated a formulation that was rather diverted from the mainstream of conventional therapy. It consisted of small silica particles (5-10 mm in diameter) covalently linked to a potent antiamoebic drug, 2-(4-aminophenoxymethyl)-5-nitro-1-methylimidazole. Silica-drug particles were injected into mice, hamsters and guinea pigs. It was found that trophozoites phagocytosed the particles in vivo and in vitro , followed by rapid cell death due to the released drug. Analysis of mouse serum revealed that no drug was absorbed from the intestine after placement of the drug-containing particles in the intestine. The antiamoebic activity of particles recovered from the intestine was almost fully retained. This novel antiamebic concept may be useful for luminal therapy for asymptomatic amebiasis and may minimize side effects and frequency of administration.
Osmotic controlled drug delivery
The OROS-CT (Alza corporation) can be used to target the drug locally to the colon for the treatment of disease or to achieve systemic absorption that is otherwise unattainable (235). The OROS-CT system can be single osmotic unit or may incorporate as many as 5-6 push-pull units (236), each 4mm in diameter, encapsulated with in a hard gelatin capsule (Figure 16). Each bilayer push pull unit contains an osmotic push layer and a drug layer, both surrounded by a semipermeable membrane. An orifice is drilled through the membrane next to the drug layer. Immediately after the OROS-CT is swallowed, the gelatin capsule containing the push-pull units dissolves. Because of its drug-impermeable enteric coating, each push-pull unit is prevented from absorbing water in the acidic aqueous environment of the stomach and hence no drug is delivered. As the unit enter the small intestine, the coating dissolve in this higher pH environment (pH >7), water enters the unit, causing the osmotic push compartment to swell and concomitantly creates a flowable gel in the drug compartment. Swelling of the osmotic push compartment forces drug gel out of the orifice at a rate precisely controlled by the rate of water transport through the semipermeable membrane. For treating ulcerative colitis, each push pull unit is designed with a 3-4 hour post gastric delay to prevent drug delivery in the small intestine. Drug release begins when the unit reaches the colon. OROS-CT units can maintain a constant release rate for up to 24 h in the colon or can deliver drug over an internal as short as 4 hour.
Figure 16: Cross section of the OROS-CT colon targeted drug delivery system.
Summary
Several systems are currently being investigated as potential means for targeting of drugs to colon. One approach is prodrug formation that requires spontaneous or enzymatic transformation within the biological environment in order to release the drug. Release of the drug from prodrug can be accomplished by utilization of some specific property at the target site such as altered pH or high activity of certain enzymes relative to nontarget tissues. Formulation can be coated with pH sensitive polymer, which dissolves at the pH of the colon. Most of the enteric polymers dissolve in the terminal ileum or at ileocaecal junction. To target the drugs specifically to colon, it is to be coated with either hydrophilic or hydrophobic polymer along with enteric polymer. At the neutral or slightly alkaline pH of the terminal ileum, the enteric coating breaks and the coating of second polymer would carry the drug to the target site. The colon is rich in harboring excellent microflora, which can be used to the target the drug release in the colon. Formulation coated with microbially degradable polymers (azo polymers) passes intact from hostile environment of upper GIT and liberates the drug after reduction and thus degradation of azo bonds by azo reductase enzymes present exclusively in the colon. Polysaccharides represent class of material that exhibits favorable properties for fabrication of colonic delivery systems. Formulation protected with polysaccharides remains intact in the adverse enzymatic environment of stomach and small intestine. Release of drug from such formulation takes place after degradation of polysaccharides due to the cleavage effect of polysaccharidases found in the colon. The most frequently encountered problem with the use of polysaccharides is their high water solubility, which causes the partial release of drugs in the upper GIT. This can be rectified by crosslinking of polysaccharides with agents such as glutaraldehyde, epichlorhydrin and sodium trimetaphosphate, which renders them hydrophobic. Multiple coated dosage form provides promising approach for the delivery of drugs to the colon. It is expected that film coated formulation composed of an enteric polymer and a hydrophilic polymer provides release of drugs at target site by delaying release in the upper GIT. All the approaches provide means for treatment of local diseases associated with the colon or for systemic absorption of poorly absorbable drugs. The need is to identify the appropriate approach, which can results in the delivery of drugs in a safe, effective and less expensive manner with minimum fluctuation in terms of release of drugs at target site.
Acknowledgements
One of the authors is thankful to University Grants Commission, New Delhi, India and Council of Scientific and Industrial Research, New Delhi, India for providing financial assistance to carry out project on colon targeting of drugs.
References
b-D-glucoside and dexamethasone in experimental inflammatory bowl disease in guinea pig. J Pharm Pharmacol, 43: 353-355, 1991.
Sarasija, S. and Hota, A., Colon-specific drug delivery systems. Ind J Pharm Sci, 62: 1-8, 2000.
Raffi, R., Franklin, W. and Cerniglia, C.E., Azoreductase activity of anaerobic bacteria isolated from human intestinal microflora. Appl Environ Mivrobiol, 56: 2146-2151, 1990.
Raffi, R., Franklin, W., Heflich R.H. and Cerniglia, C.E., Reduction of nitroaromatic compounds by anaerobic bacteria isolated from the human intestinal tract. Appl Environ Mivrobiol, 57: 962-968, 1991.
Walker, R. and Ryan, A.J., Some molecular parameters influencing rate of reduction of azo compounds by intestinal microflora. Xenobiotica, 1: 483-486, 1971.
Azad Khan, A.K., Truelove, S.C. and Aronseq J.K., the disposition and metabolism of sulphasalazine (salicylazosulphapyridine) in man. Br J Clin Pharmacol, 13: 523-528, 1982.
Chan, R.P., Pope, D.J., Gilbert, A.P.Baron, J.H. and Lennard-Jones, J.P., Studies of two novel sulfaslazine analogue: ipsalazine and balsalazine. Dig Dis Sci, 28: 609-615, 1983.
Hartalsky, A., Salicylazobenzoic acid in ulcerative colitis. Lancet, 1: 960, 1982.
Garreto, M., Ridell, R.H. and Wurans, C.S., Treatment of chronic ulcerative colitis with poly-ASA: A new nonabosrbable carrier for release of 5-aminosalicylic acid in the colon. Gastroenterology, 84: 1162, 1981.
Willoughby, C.P., Aronson, J.K., Agback, H., Bodin, N.O., Anderson, E. and Truelove, S.C., Disposition in normal volunteers of sodium azodisalicylate, a potential therapeutic agent in inflammatory bowl disease. Gut, 22: A431, 1981.
Willoughby, C.P., Aronson, J.K., Agback, H., Bodin, N.O., Anderson, E. and Truelove, S.C., Disposition in normal volunteers of sodium azodisalicylate, a potential therapeutic agent in ulcerative colitis. Gut, 23: 1081-1087, 1982.
Lauristein, K., Hansen, J., Ryde, H.M. and Rask-Madsen, J., Colonic azodisalicylate metabolism determined by in vivo dialysis in healthy volunteers and patients with ulcerative colitis. Gastroenterology, 86: 1496-1500, 1984.
Sandberg-Gertzen, H., Ryde, H.M. and Janerot, G., Absorption, metabolism and excretion of a single 1 g dose of azodisal sodium in subjects with ileostom. Scan J Gastroenterol, 88: 107-111, 1983.
Van Hogezand, R.A., Van Hees, P.A.M., Zwaneburg, B., Van Rossum, J.M. and Van Tongeren, J.H.M. Disposition of sodium azodisalicylate in healthy subjects, Gastroenterolgy, 88: 717-722, 1985.
Rao, S.S.C., Read, N.W. and Holdsworth, C.D. Influence of olsalazine on gastrointestinal transit in ulcerative colitis. Gut, 28: 1474-1477, 1987.
Pamucku, R., Hanauer, S. and Chang, E.B., Effect of disodium azosalicylate on electrolyte transport in the rabbit ileum and colon in vitro. Gastroenterology, 95: 975-981, 1988.
McIntyre, P.B., Rodrigues, C.A. and Lennard-Jones, J.E., Balsalazine in the maintenance treatment of patients with ulcerative colitis, a double-blind comparison with sulphasalazine. Aliment Pharmacol Ther, 2: 237-243, 188.
Riley, S.A. and Turnberg, L.A., Sulphasalazine and the aminosalicylates in the treatment of inflammatory bowl disease. Q J Med, 75: 561-562, 1990.
Klotz, U., Clinical pharmacokinetics of sulphasalazine, its metabolites and other prodrugs of 5-aminosalicylic acid. Clin Pharmacokint, 10: 285-302, 1985.
Friendman, G., Sulphasalazine and new analogues. Am J Gastroenterol, 81: 141-144, 1986.
Garretto, M., Riddell, R.H. and Winans, C.S., Treatment of ulcerative colitis with poly-ASA: a new non-absorbable carrier for release of 5-aminosalicylic acid release in vitro. Gasroenterology, 16: 211-212, 1989.
Sakuma, S., Lu, Zheng-Rong, Kopeková, P. and Kopeek, J., Biorecognizable HPMA copolymer-drug conjugates for colon-specific delivery of 9-aminocamptothecin. J Control Rel, 75: 365-379, 2001.
Friend, D.R., Glycosides in colonic drug delivery. In: Friend, D.R. (Ed). Oral colon specific drug delivery. CRC Press, Boca Raton, pp 153-187, 1992.
Pownall, J.H., Hickson, D.L. and Smith, L.C., Transport of Biological Lipophils: Effect of lipophilic structure. J Am Chem Soc, 105: 2440-2445, 1983.
Kobashi, K., Nishimura, T., Kusaka, M., Hottari, M. and Namba, T., Metabolism of sennoids by human intetinal bacteria. J Med Plant Res, 40: 225-236, 1980.
Conchie, J. and Macleod, D.C., Glycosidase in mammalian alimentary tract. Nature, 184: 1233, 1959.
Rubinstein, A., Approaches and opportunities in colon-specific drug delivery. Crit Rev Ther Carr Syst, 12: 101-149, 1995.
Friend, D.R. and Chang, G.W., A colon-specific drug delivery based on the drug glycosidases of colonic bacteria. J Med Chem, 27: 261-266, 1984.
Friend, D.R. and Chang, G.W., Drug Glycosides: Potential prodrug for colon-specific drug delivery. J Med Chem, 85: 51-57, 1985.
Friend, D.R. and Tozer, T.N., Drug glycoside in colon-specific drug delivery. J Control Rel, 19: 109-120, 192.
Friend, D.R., Philips, S, McLeod, A and Tozer, T.N., Relative anti-inflammatory effect of oral dexmamethasone-
Minami, K., Hiramaya, F. and Uekama, K., Colon-specific drug delivery based on cyclodextrin prodrug: release behaviour of biphenyl acetic acid form its cyclodextrin conjugates in rat intestinal tract after oral administration. J Pharm Sci, 87: 715-720, 1998.
Scheline, R.P., Drug metabolism by intestinal microorganism. J Pharm Sci, 57: 2021-2037, 1968.
Simpkins, J.W., Smulkowski, M., Dixon, R. and Tuttle, R., Evidence for the delivery of narcotic antagonists to the colon as their glucuronide conjugates. J Pharmacol Exp Ther, 244: 195-205, 1988.
b-D-glucuronide for colon-specific drug delivery. Pharm Res, 10: 1553-1562, 1993. Haeberlin, B., Rubas, W., Nolen, II, H.W. and Friend, D.R., In vitro evaluation of dexamethasone-
Mcleod, A.D., Feclorak, R.N., Friend, D.R., Tozer, T.N. and Cui, N., A glucocorticoid prodrug facilitates normal mucosal function in rat colitis without adrenal suppression. Gastroenterology, 106: 405-413, 1994.
Cui, N. Friend, D.R. and Fedorak, R.N., A Budesonide prodrug accelerates treatment of colitis in rats. Gut, 35: 1439-1446, 1994.
Fedorak, R.N., Haeberlin, B. and Empey, L.R., Colonic drug delivery of dexamethasone from a prodrug accelerates healing of colitis in rat without adrenal suppression. Gastroenterology, 108: 1688-1699, 1995.
Nolen, H.W. III, Fedorak, R.N. and Friend, D.R., Steady-state pharmacokinetics of corticosteroid delivery from glucuronide prodrugs in normal and colitic rat. Biopharm Drug Dispos, 18: 681-695, 1997.
b-cyclodextrin: properties and usage in pharmaceutical formulations. Pharm Ztg Wiss, 4: 5-10, 1991. Loftsson, T., Brewster, M.E., Derendorf, H. and Bodor, N., 2-hydroxypropyl-
Uekama, K., Hiramaya, F. and Irie, T., Pharmaceutical uses of cyclodextrin derivatives. In: (Szycher, M., Ed.). High performance Biomaterials, Technomic publishing, Lancaster, pp 789-806, 1991.
Szejtli, J. and Szente, L. Ed. Proceedings of the Eighth international symposium on cyclodextrins, Kluwer, Dordrecht, 1996.
Stella, V.J. and Rajewski, R.A., Cyclodextrins: their future in drug formulation and delivery. Pharm Res, 14: 556-567, 1997.
Andersen, G.H., Robbins, F.M., Domingues, F.J., Morres, R.G. and Long, C.L., The utilisation of schardinger dextrins by the rat. Toxicol Appl Pharmacol, 5: 257-266, 1983.
a and b-cyclodextrins by bacteroids of the human colon. J Agric Food Chem, 32: 1316-1321, 1984. Antenucci, R. and Palmer, J.K., Enzymatic degradation of
b-cyclodextrin in rat. Arzneim Forsch / Drug Research, 35: 1042-1047, 1985. Gerloczy, A., Fonagy, A. Keresztes, P., Perlaky, L. and Szejtli, J., Absorption, distribution, excretion and metabolism of orally administered 14C-
b-cyclodextrin in the human intestine. J Nutr, 123: 676-680, 1993. Flourie, B., Molis, C. Achour, L., Dupas, H., Hatat, C. and Rambaud, J.C., Fate of
Karunaratne, D.N., Farmer, S. and Hancock, R.E.W., Synthesis of bulky beta-lactams for inhibition of cell surface beta-lactamase activity. Bioconjug Chem, 4: 434-439, 1993.
Tanaka, H., Kominato, K. and Yamamoto, R., Synthesis of doxorubicin-cyclodextrin connjugates. J Antibio, 47: 1025-1029, 1194.
Coates, J.H., Easton, C.J., Fryer, N.L. and Lincolin, S.F., Complementary diastereo selectivity in the synthesis and hydrolysis of acylated cyclodextrins. Chem Lett, 1153-1156, 1994.
Minami, K., Hiramaya, F. and Uekama, K., Colon-specific drug delivery based on a cyclodextrin prodrug: release behaviour of biphenyl acetic acid from its cyclodextrin conjugates in rat intestinal tracts after oral administration. J Pharm Sci, 87: 715-720, 1998.
Uekama, K., Minami, K. and Hirayama, F., 6A-O-[(4-biphenylyl)acetyl]-alpha-, -beta- and -gamma-cyclodextrins and 6A-deoxy-6A-[[(4-biphenylyl)acetyl]amino]-alpha-, -beta-, and -gamma-cyclodextrins: potential prodrugs for colon-specific delivery. J Med Chem, 40:2755-61, 1997.
Hiramaya, F., Minami, K., and Uekama, K., In vitro evaluation of biphenyl acetic acid-beta-cyclodextrin conjugates as colon-targeting prodrugs: Drug release behaviour in rat biological media. J Pharm Pharmacol, 48: 27-31, 1996.
Yano, H., Hirayama, F., Kamada, M., Arima, H., Uekama, K. and Uekama, K., Colon-specific delivery of prednisolone-appended alpha-cyclodextrin conjugate: alleviation of systemic side effect after oral administration. J Control Rel, 79: 103-12, 2002.
a-, b- and g-cyclodextrins: substitution at secondary hydroxyl groups and in vitro hydrolysis behaviour. J Pharm Sci, 90: 493-503, 2001. Yano, H., Hiramaya, F., Arima, H. and Uekama, K., Preparation of prednisolone-appended
Frank, D.W., Gray, J.E. and Weaver, R.N., Cyclodextrin nephrosis in the rat. Am J Pathol, 83: 367-382, 1976.
b-cyclodextrin derivatives. J Pharm Sci, 84: 927-932, 1995. Rajeskwi, R.A., Traiger, G., Bresnahan, A., Jaberaboansari, P., Stella, V.J. and Thompson, D.O., Preliminary safety evaluation of parenterally administered sulfoalkyl ether
Gerloczy, A., Antal, S., Szatmari, I., Muller-Horvath, R. and Szejti, J., Mins 5th Int Symp cyclodextrins. Duchene, D. (Ed.) Editions de Sante: Paris, 507-513, 1990.
Monbaliu, J., Van Beijsterveldt, L., Meuldermans, W., Szathnari, S. and Heykants, J., Mins 5th Int Symp cyclodextrins. Duchene, D. (Ed.) Editions de Sante: Paris, 514-517, 1990.
b-cyclodextrins as novel sustained release carries for a water soluble drug, molsidomine. J Pharm Pharmacol, 46: 714-717, 1994. Uekama, M., Horikawa, T., Yamanaka, M. and Hiramaya, F., Peracylated
Larsen, C., Harboe, E., Johansen, M. and Olesen, H.P., Macromoleculer prodrugs. XVI. Colon targeted delivery. Comparison of the rate of release of naproxen from dextran ester prodrug in homogenates of various segments of the pig gastrointestinal tract. Pharm Res, 6: 995-999, 1989.
Harboe, E., Larsen, C., Johansen, M. and Olesen, H.P., Macromoleculer prodrugs. XV. Colon targeted delivery. Bioavailability of naproxen from orally administered dextran ester prodrugs varying molecular size in the pig. Pharm Res, 6: 919-923, 1989.
Harboe, E., Larsen, C., Johansen, M. and Olesen, H.P., Macromoleculer prodrugs. XIV. Absorption characteristics of naproxen after oral administration of a dextran T-70-naproxen ester prodrugs in pigs. Int J Pharm, 53: 157-165, 1989.
Johansen, M. and Larsen, C., Stability kinetics and of hydrolysis of metronidazole monosuccinate in aqueous solution and in plasma. Int J Pharm, 21: 201-209, 1984.
Johansen, M. and Larsen, C., A comparison of the chemical stability and the enzymatic hydrolysis of a series of aliphatic and aromatic ester derivatives of metronidazole. Int J Pharm, 27: 219-231, 1985.
Vermeersch, J., Vandoorne, F., Permentier, D. and Schacht, E., Macromoleculer prodrugs of metronidazole 1. Esterification of hydroxyl containing Polymers with metronidazole monosuccinate. Bull Soc Chim Belg, 94: 591-596, 1985.
McLeod, A.D., Friend, D.R. and Tozer, T.N., Synthesis and chemical stability of glucocorticoid-dextran ester: Potential prodrugs for colon-specific drug delivery. Int J Pharm, 92: 105-114, 1993.
McLeod, A.D., Tolentino, L. and Tozer, T.N., Glucocorticoid-dextran ester conjugates as Potential prodrugs for colon-specific drug delivery: Steady-state pharmacokinetics in rat. Biopharm Drug Dispos, 15: 151-164, 1994.
McLeod, A.D., Fedorak, R.N., Friend, D.R., Tozer, T.N. and Cui, N., A glucocorticoid prodrug facilitates normal mucosal function in rat colitis without adrenal suppression. Gastroenterology, 106; 405-413, 1994.
Nakamura, J., Asai, K., Nishida, K. and Sasaki, H., A novel prodrug of salicylic acid, salicylic acid-glutamic acid conjugate utilising hydrolysis in rabbit intestinal microorganism. Chem Pharm Bull, 40: 2164-2168, 1992.
Nakamura, J., Kido, M., Nishida, K. and Sasaki, H., Hydrolysis of salicylic acid-tyrosine salicylic acid-methionine prodrugs in rabbits. Int J Pharm, 87: 59-66, 1992.
Nakamura, J., Kido, M., Nishida, K. and Sasaki, H., Effect of oral pretreatment with antibiotics on the hydrolysis of salicylic acid-tyrosine and salicylic acid-methionine prodrugs in rabbit intestinal microorganisms. Chem Pharm Bull, 40: 2572-2576, 1992.
Nakamura, J., Tagami, K., Nishida, K. and Sasaki, H., Unequal hydrolysis of salicylic acid-D-alanine and salicylic acid-L-alanine conjugate in rabbit intestinal microorganisms. Chem Pharm Bull, 40: 547-549, 1992.
Schacht, E., Gevaert, A. and Kenawy, E.R., Polymers for colon specific drug delivery. J Control Rel, 39: 327-328, 1996.
Schacht, E., Callant D. and Verstraete, W., Synthesis and evaluation of polymeric prodrugs of 5-aminosalicylic acid. Proc Int Symp Contr Rel Bioact Materl, 18: 686-687, 1991.
Leopold, C.S. and Friend, D.R., In vitro study for the assessment of poly(L-Aspartic acid) as a drug carrier for colon-specific drug delivery. Int J Pharm, 126: 139-145, 1995.
Leopold, C.S. and Friend, D.R., In vivo pharmacokinetics study for the assessment of poly(L-Aspartic acid) as a drug carrier for colon-specific drug delivery. J Pharmacokinet Biopharm, 23: 397-406, 1995.
Wilding, I.R., Hardy, J.G., Sparrow, R.A., Davis, S.S, Daly, P.B. and English, J.R., In vivo evaluation of enteric coated naproxan tablets using gamma scintigraphy. Pharm Res, 9: 1436-1441, 1992.
Khan, M.Z.I., Dissolution testing for sustained or controlled release oral dosage forms and correlation with in vivo date: challenges and opportunities. Int J Pharm, 140: 141-143, 1996.
Dressman, J.B., Amidon, C., Reppas, C. and Shah, V.P., Dissolution testing as a prognostic tool for oral drug absorption: Immediate release dosage forms, Pharm Res, 15: 11-22, 1998.
Ashford, M., Fell, J.T., Attwood, D. and Woodhead, P.J., An in vitro investigation into the suitability of pH-dependent polymers for colon targeting. Int J Pharm, 91: 241-243, 1993.
Spitael, J. and Kinget, R., Solubility and dissolution rate of enteric polymers. Acta Pharm Technol, 25: 163-168, 1979.
Spitael, J. and Kinget, R., Factors affecting the dissolution rate of enteric coatings. Pharm Ind, 39: 502-505, 1977.
Spitael, J., Kinget, R. and Naessens, K., Dissolution rate of cellulose acetate phthalate and Bronsted catalysis. Pharm Ind, 42: 846-849, 1980.
Spitael, J. and Kinget, R., Influence of solvent composition upon film-coating. Pharm Acta Helv, 55: 157-160, 1980.
Touitou, E. and Rubinstein, A., Targeted eternal delivery of insulin to rats. Int J Pharm, 30: 95-99, 1986.
Thomos, P., Richards, D. and Richards, A., Absorption of delayed release prednisolone in ulcerative colitis and Chron's disease. J Pharm Pharmacol, 37: 757-758, 1985.
Van Saene, J.J.M., Van Saene, H.F.K., Geitz, J.N., Tarko-Smit, N.J.P. and Lerk, C.F., Quinolones and colonization resistance in human volunteers, Pharm Weekbl Sci Ed, 8: 67-71, 1986.
Azad Khan, K.A., Piris, J. and Truelove, S.C., An experiment to determine the active therapeutic moiety of sulphasalazine. Lancet, 2: 892-895, 1977.
Bogentoft, C., Eskilsson, C., Jonsson, U.E., Lagerstorm, P.O., Lovgren, K. and Rosen, L., Delivery of drug to the colon by means of a new microencapsulated oral dosage form. Acta Phrarm Suec, 20: 311-314, 1983.
Watkinson, G., Sulphasalazine: A review of 40 years experience. Drugs, 32(suppl 1): 1-11, 1986.
Riley, S.A., Lecarpentier, J., Mani, V., Goodman, M.J., Mandal, B.K. and Turnberg, L.A., Sulphasalazine induced seminal abnormalities in ulcerative colitis: Results of mesalazine substitute. Gut, 28: 1008-1012, 1987.
Al Mardini, H., Lindsay, D.C., Deighton, C.M. and Record, C.A., Effect of polymer coating on faecal recovery of ingested 5-aminosalicylic acid in patients with ulcerative colitis. Gut, 28: 1084-1089, 1987.
Kim, C.K., Shin, H.J., Yang, S.G., Kim, J.H. and Oh, Y., Once a day oral dosing regimen of cycloscoporin A: Combined therapy of cycloscoporin a premicroemulsion concentrates and enteric coated solid-state premicroemulsion concentrates, Pharm Res, 18: 454-459, 2001.
Levine, D.S., Raisys, V.A. and Ainardi, V., Coating of oral beclomethasone dipropionate capsules with cellulose acetate phthalate enhances delivery of topically active anti inflammatory drug to the terminal ileum. Gastroenterology, 92: 1037-1044, 1987.
Hardy, J.G., Evans, D.F., Zaki, I., Clark, A.G., Tonnesen, H.H. and Gamst, O.N., Evaluation of an enteric coated naproxen tablet using gamma scintigraphy and pH monitoring. Int J Pharm, 37: 245-250, 1987.
Khan, M.Z., Prebeg, Z. and Kurjakovic, N., A pH-dependent colon targeted oral drug delivery system using methacrylic acid copolymers I. Manipulation of drug release using Eudragit® L100-55 and Eudragit® S100 combinations. J Control Rel, 58: 215-222, 1999.
Lorenzo-Lamosa, M.L., Remunan-Lopez, C., Vila-Jato, J.L. and Alonso, M.J., Design of microencapsulated chitosan microspheres for colonic drug delivery. J Control Rel, 52: 109-118, 1998.
Dew, M.J., Ryder, R.E.J., Evans, N., Evans, B.K. and Rhodes, J., Colonic release of 5-aminosalicylic acid from an oral preparation in active ulcerative colitis. Br J Clin Pharmcol, 16: 185-187, 1983.
Dew, M.J., Ebden, P., Kidwai, S., Lee, G., Evans, B.K. and Rhodes, J., Comparison of the absorption and metabolism of sulphasalazine and acrylic-coated 5-aminosalicylic acid in normal subjects and in patients with colitis. Br J Clin Pharmcol, 17: 474-476, 1984.
Hardy, J.G., Healy, J..N.C. and Reynolds, J.R., Evaluation of an enteric coated delayed release 5-aminosalicylic acid tablet in patients with inflammatory bowl disease. Aliment Pharmacol Ther, 1: 273-280, 1987.
Schroeder, K.W., Tremaine, W.J. and Ilstrup, D.M., Coated oral 5-aminosalicylic acid therapy for mild to moderately active ulcerative colitis. N Engl J Med, 317: 1625-1629, 1987.
Norlander, B., Gotthard, R. and Storm, M., Pharmacokinetics of 5-aminosalicylic acid enteric-coated tablet and suppository dosage form. Aliment Pharmacol Ther, 3: 333-342, 1989.
Jarnerot, G., Newer 5-aminosalicylic acid based drugs in chronic inflammatory bowel disease. Drugs, 37: 73-86, 1989.
Faber, S.M. and Korelitz, B.I., Experience with Eudragit-S-Coated mesalamine (Asacol) in inflammatory bowl disease: An open study. J Clin Gastroenterol, 17: 213-218, 1993.
Morishita, I., Morishita, M., Takayama, K., Machida, Y. and Nagai, T., Enteral insulin delivery by microspheres in three different formulations using Eudragit L100 and S100. Int J Pharm, 91: 29-37, 1993.
Cole, E.T., Scott, R.A., Connor, A.L., Wilding, I.R., Petereit, H.U., Schminke, C., Beckert, T. and Cade, D., Enteric coated HPMC capsules designed to achieve intestinal targeting. Int J Pharm, 231: 83-95, 2002.
Peeters, R. and Kinget, R., Film-forming polymers for colonic drug delivery. I. Synthesis and physical and chemical properties of methyl derivatives of Eudragit S. Int J Pharm, 94: 125-134, 1993.
Rodríguez, M., Vila-Jato, J.L. and Torres, D., Design of a new multiparticulate system for potential site-specific and controlled drug delivery to the colonic region. J Control Rel, 55: 67-77, 1998.
Ashford, M., Fell, J.T., Attwood, D., Sharma, H. and Woodhead, P.J., An in vivo investigation into the suitability of pH-dependent polymers for colonic targeting. Int J Pharm, 95: 193-199, 1993.
Rodriguez, M., Antúnez, J.A., Taboada, C., Seijo, B. and Torres, D., Colon-specific delivery of Budesonide from microencapsulated cellulosic cores: evaluation of the efficacy against colonic inflammation in rats. J Pharm Pharmacol, 53: 1207-15, 2001.
Markus, W. Rudolph, Klein, S., Beckert, T.E., Petereit, H. and Dressman, J.B., A new 5-aminosalicylic acid multi-unit dosage form for the therapy of ulcerative colitis Eur J Pharm Biopharm, 51: 183-190, 2001.
Saffran, M., Kumar, G.S., Savariar, C., Burnham, J.C., Williams, F. and Neckers, D.C., A new approach to the oral administration of insulin and other peptide drugs. Science, 233: 1081-1084, 1986.
Saffran, M., Bedra, C., Kumar, G.S. and Neckers, D.C., Vasopressin: a new model for the study of effects of additives on the oral and rectal administration of peptide drugs. J Pharm Sci, 77: 33-38, 1988.
Saffran, M., Bedra, C., Kumar, G.S., Neckers, D.C., Pena, J., Jones, R.H. and Field, J.B., Biodegradable azo polymer coating for oral delivery of peptide drugs. Biochem Soc Trans, 18: 752-754, 1990.
Saffran, M., Field, J.B., Pena, J., Jones, R.H. and Okuda, Y., Oral insulin in diabetic dogs. J Endocrinol, 131: 267-278, 1991.
Van den Mooter, G., Samyn, C. and Kinget, R., Azo polymers for colon-specific drug delivery. Int J Pharm, 87: 37-46, 1992.
Van den Mooter, G., Samyn, C. and Kinget, R., Azo polymers for colon-specific drug delivery. Part II: Influence of the type of azo polymer on the degradation by intestinal microflora. Int J Pharm, 97: 133-139, 1993.
Kinget, R., Kalala, W., Vervoort, L. and Van den Mooter, G., Colonic drug targeting. J Drug Target, 6:129-149, 1998.
Van den Mooter, G., Samyn, C. and Kinget, R., Characterisation of colon-specific azo polymers: A study of the swelling properties and the permeability of isolated polymer films. Int J Pharm, 11: 127-136, 1994.
Van den Mooter, G., Samyn, C. and Kinget, R., In vivo evaluation of a colon-specific drug delivery system: An absorption study of theophylline from capsules coated with azo polymers in rat. Pharm Res, 12: 244-247, 1995.
Kalala, W., Kinget, R., Van den Mooter, G. and Samyn, C., Colonic drug-targeting: in vitro release of ibuprofen from capsules coated with poly(ether-ester) azopolymers. Int J Pharm, 139: 187-195, 1996.
Symn, C., Kala, W., Van den Mooter, G. and Kinget, R., Synthesis and in vitro biodegradation of poly(ether-ester) azo polymers designed for colon targeting. Int J Pharm, 121: 211-216, 1995.
Kimura, Y., Makita, Y., Kumagi, T., Yamane, T., Kiato, H., Sasatani, H. and Kim, S.I., Degradation of azo-containg polyurethane by the action of intestinal flora: its metabolism and application as a drug delivery system. Polymer, 33: 5294-5299, 1992.
Ueda, T., Yamaoka, M., Miyamoto, M. and Kimura, Y., Bacterial reduction of azo compounds as a model reaction for the degradation of azo-containing polyurethane by the action of intestinal flora. Bull Chem Soc Jpn, 69: 1139-1142, 1996.
Yamaoka, T., Makita, Y., Sasatani, H., Kim, S.I. and Kimura, Y., Linear type azo-containing polyurethane as drug-coating material for colon-specific delivery: its properties, degradation behaviour, and utilization for drug formulation. J Control Rel, 66: 187-97, 2000.
Chavan, M.S., Sant, V.P. and Nagarsenker, M.S., Azo-containing urethane analogues for colonic drug delivery: synthesis, characterization and in-vitro evaluation. J Pharm Pharmacol, 53: 895-900, 2001.
Lehmann, K.O.R. and Drehher, Methacrylated galactomannan coating for colon-specific drug delivery. Proc Int Symp Contrl Rel Bioact Mater, 18: 331-332, 1991.
Vervoort, L. and Kinget, R., In vitro degradation by colonic bacteria of inulin HP incorporated in Eudragit RS films. Int J Pharm, 129: 185-190, 1996.
Wakerly, Z., Fell, J.T., Attwood, D. and Parkins, D., Pectin/ethyl cellulose film coating formulation for colonic drug delivery. Pharm Res, 13: 1210-1212, 1996.
Brondsted, H., Andersen, C. and Hovgaard, L., Crosslinked dextran - a new capsule material for colon targeting of drugs. J Control Rel, 53: 7-13, 1998.
Bauer, K.H. and Kesselhut, J.F., Novel pharmaceutical excipients for colon targeting. STP Pharm Sci, 5: 54-59, 1995.
Hirsch, S., Binder, V., Kolter, K., Kesselhut, J.F., and Bauer, K.H., Lauroyldextrin as a coating material for site-specific drug delivery to the colon: In vitro dissolution of coated tablets. Proc Int Symp Contrl Rel Bioact Mater, 24: 379-380, 1997.
Lamprecht, A., Torres, H.R., Schafer, U. and Lehr, C.M., Biodegradable microparticles as a two-drug controlled release formulation: a potential treatment of inflammatory bowel disease, J Control Rel, 69: 445-454, 2000.
Rubinstein, A., Radai, R., Ezra, M., Pathak, S. and Rokem, J.M., In vitro evaluation of calcium pectinate: A potential colon-specific drug delivery carrier. Pharm Res, 10: 258-263, 1993.
Rubinstein, A. and Radai, R., Pectic salt as a colonic delivery system. Proc Int Symp Contrl Rel Bioact Mater, 18: 221-222, 1991.
Ashford, M., Fell, J.T., Attwood, D., Sharma, H. and Woodhead, P., Studies on pectin formulations for colonic drug delivery. J Control Rel, 30: 225-232, 1994.
Wakerly, Z., Fell, J.T., Attwood, D. and Parkins, D., Studies on drug release from pectin/ethylcellulose film-coated tablets: a potential colonic delivery system. Int J Pharm, 153: 219-224, 1997.
Macleod, G.S., Fell, J.T. and Collett, J.H., Studies on the physical properties of mixed pectin/ethylcellulose films intended for colonic drug delivery. Int J Pharm, 157: 53-60, 1997.
Fernandez-Hervas, M.J. and Fell, J.T., Pectin/chitosan mixtures as coatings for colon-specific drug delivery: an in vitro evaluation. Int J Pharm, 169: 115-119, 1998.
Macleod, G.S., Fell, J.T., Collett, J.H., Sharma, J.L. and Smith, A.M., Selective drug delivery to the colon using pectin: chitosan: hydyroxypropyl methylcellulose film coated tablets. Int J Pharm, 187: 251-257, 1999.
Turkoglu, M., Ugurlu, T., In vitro evaluation of pectin-HPMC compression coated 5-aminosalicylic acid tablets for colonic delivery. Eur J Pharm Biopharm, 53: 65-73, 2002.
Semde, R., Amighi, K., Devleeschouwer, M.J. and Moes, A.J., Effect of pectinolytic enzymes on the theophylline release from pellets coated with water insoluble polymers containing pectin HM or calcium pectinate. Int J Pharm, 197:169-179, 2000.
Semde, R., Amighi, K., Devleeschouwer, M.J. and Moes, A.J., Studies of pectin HM/Eudragit RL/Eudragit NE film-coating formulations intended for colonic drug delivery. Int J Pharm, 197: 181-92, 2000.
Munjeri, O., Collett, J.H. and Fell, J.T., Hydrogel beads based on amidated pectins for colon-specific drug delivery: the role of chitosan in modifying drug release. J Control Rel, 46: 273-278, 1997.
Sriamornsak, P. and Nunthanid, J., Calcium pectinate gel beads for controlled release drug delivery: I. Preparation and in vitro release studies. Int J Pharm, 160: 207-212, 1998.
El-Gibaly, I., Oral delayed-release system based on Zn-pectinate gel (ZPG) microparticles as an alternative carrier to calcium pectinate beads for colonic drug delivery. Int J Pharm, 232: 199-211, 2002.
Grahan, N.B., McNeil, M.E., Hydrogels for controlled drug delivery, Biomaterials, 5: 27-36, 1984.
Bae, Y.H. and Kim, S.W., Hydrogels delivery systems based on polymer blends, block-copolymers or interpenetrating networks, Adv Drug Del Rev, 11: 109-135, 1993.
Kopecek, J., Kopeckova, P. and Brondsted, H., Polymers for colon-specific oral drug delivery. J Control Rel, 19: 121-130, 1992.
Shanta, K.L., Ravichandran, P. and Rao, K.P., Azo polymeric hydrogels for colon targeted drug delivery. Biomaterials, 16: 1313-1318, 1995.
Brondsted, H., Kopeckova, P., Hydrogels for site-specific oral drug delivery: Synthesis and characterisation. Biomaterials, 12: 584-592, 1991.
Brondsted, H. and Kopecek, J., Hydrogels for site specific drug delivery to colon: in vitro and in vivo degradation, Pharm Res, 9: 1540-1545, 1992.
Ghandehari, H., Kopecekova, P. and Kopecek, J., In vitro degradation of pH-sensitive hydrogels containing aromatic azo bonds. Biomaterials, 18: 861-72, 1997.
Ulbrich, K., Strohalm, J. and Kopecek, J., Polymers containing enzymatically degradable bonds. VI. Hydrophilic gels cleavable by chymotrypsin. Biomaterials, 3: 150-154, 1982.
Vervoort, L., Rombaut, P., Van den Mooter, G., Augustijns, P. and Kinget, R., Inulin hydrogels. II. In vitro degradation study. Int J Pharm, 172: 137-145, 1998.
Vervoort, L., Rombaut, P., Van den Mooter, G., Augustijns, P. and Kinget, R., Inulin hydrogels. I. Dynamic and equilibrium swelling properties. Int J Pharm, 172: 127-135, 1998.
Maris, B., Verheyden,L., Reeth, K.V., Samyn, C., Augustijns, P., Kinget, R. and Van den Mooter, G., Synthesis and characterisation of inulin-azo hydrogels designed for colon targeting. Int J Pharm, 213: 143 - 152, 2001.
Vervoort, L., Van den Mooter, G., Augustijns, P., Busson, R., Toppet, S. and Kinget, R., Inulin hydrogels as carrier for colonic drug targeting :I. Synthesis and characterization of methacrylated inulin and hydrogel formation. Pharm Res, 14: 1730-1736, 1997.
Stubbe, B., Maris, B., Van den Mooter, G., and Demeester, J., The in vitro evaluation of azo containing polysaccharide gels' for colon delivery. J Control Rel, 75: 103-114, 2001.
Hovgaard, L. and Brondsted, H., Dextran hydrogels for colon-specific drug delivery. J Control Rel, 36: 159-166, 1995.
Chiu, H.C., Hsiue, G.H., Lee, Y.P. and Huang, L.W., Synthesis and characterization of pH-sensitive dextran hydrogels as a potential colon-specific drug delivery system. J Biomater Sci Polym Ed, 10: 591-608, 1999.
Orienti, T., and Zecchi, V., Hydrogels formed by cross-linked polyvinylalcohol as colon-specific drug delivery systems. Drug Dev Ind Pharm, 27: 877-84, 2001.
Sintov, A., Ankol, S., Levy, D. and Rubinstein, A., Enzymatic cleavage of disaccharide side groups in insoluble synthetic polymers: a new method for specific delivery of drugs to the colon. Biomaterials, 14: 483-490, 1993.
French, D.L. and Mauger, J.W., Evaluation of the physiochemical properties and dissolution characteristics of mesalamine: Relevance to controlled intestinal drug delivery. Pharm Res, 10: 1285-1290, 1993.
Cen, M., Uzun, C. and Güven, O., Controlled release of terbinafine hydrochloride from pH sensitive poly(acrylamide/ maleic acid) hydrogels Int J Pharm, 203: 149-157, 2000.
Goldsten, A..M., Alter, E..N. and Seaman, J.K., Guar gum, In: Whistler R.L. (Ed.). Oral colon specific drug delivery and their derivatives, Academic press, Florida, pp. 1-43, 1992.
Johnson, J.C. and Gee, J..M., Effect of gel forming gum on the intestinal unstirred layer and sugar transport in vitro. Gut, 22: 398-403, 1981.
Tomlin, B.J., Read, N.W., Edwards, C..A. and Duerden, B.I., The degradation of guar gum by fecal incubation system. Brit J Nutrn, 55: 481-86, 1986.
Gliko-Kabir, I., Yagen, B., Penhasi, A. and Rubinstein, A., Phosphated crosslinked guar for colon-specific drug delivery. I. Preparation and physicochemical characterization. J Control Rel, 63: 121-127, 2000.
Gliko-Kabir, I., Yagen, B., Baluom, M. and Rubinstein, A., Phosphated crosslinked guar for colon-specific drug delivery. II. In vitro and in vivo evaluation, J Control Rel, 63: 129-134, 2000.
Rama Prasad, Y.V., Krishnaiah, Y.S.R. and Satyanarayana, S., In vitro evaluation of guar gum as a carrier for colon-specific drug delivery. J Control Rel, 51: 281-287, 1998.
Gliko-Kabir, I., Yagen, B., Penhasi, A. and Rubinstein, A., Low swelling, crosslinked guar and its potential use as colon-specific drug carrier. Pharm Res, 15: 1019-1025, 1998.
Krishnaiah Y.S.R., Seetha Devi, A, Nageswara Rao, L, Bhaskar Reddy, P.R., Karthikeyan. R.S., Satyanarayana, V., Guar gum as a carrier for colon specific delivery; influence of metronidazole and tinidazole on in vitro release of albendazole from guar gum matrix tablets. J Pharm Pharmaceut Sci, 4: 235-243, 2001.
Krishnaiah, Y.S.R., Veer Raju, P., Dinesh Kumar, B., Bhaskar, P. and Satyanarayana, V., Development of colon targeted drug delivery systems for mebendazole. J Control Rel, 77: 87-95, 2001.
Krishnaiah, Y.S.R., Satyanarayana, S., Rama Prasad, Y.V. and Narasimha Rao, S., Gamma scintigraphic studies on guar gum matrix tablets for colonic drug delivery in healthy subjects. J Control Rel, 55: 245-252, 1998.
Milojevic, S., Newton, J.M., Cummings, J.H., Gibson, G.R., Botham, R.L., Ring, S.C., Stockham, M. and Allwood, M.C., Amylose as a coating for drug delivery the colon: Preparation and in vitro evaluation using 5-aminosalicylic acid pellets. J Control Rel, 38: 75-84, 1996.
Milojevic, S., Newton, J.M., Cummings, J.H., Gibson, G.R., Botham, R.L., Ring, S.C., Stockham, M. and Allwood, M.C., Amylose as a coating for drug delivery the colon: Preparation and in vitro evaluation using glucose pellets. J Control Rel, 38: 85-94, 1996.
Lenaerts V., Dumoulin Y. and Mateescu M. A., Controlled release of theophylline from crosslinked amylose tablets. J Control Rel, 15: 39-46, 1991.
Cummings, J.H., Milojevic, S., Harding, M., Coward, W.A., Gibson, G.R., Botham, R.L., Ring, S.G., Wraight, E.P., Stockham, M.A., Allwood, M.C. and Newton, J.M., In vivo studies of amylose- and ethylcellulose-coated [13C]glucose microspheres as a model for drug delivery to the colon. J Control Rel, 40: 123-131, 1996.
Salyers, A.A., Energy sources of major ntestinal fermentative anaerobes. Am J Clin Nutr, 32: 158-163, 1979.
Rubinstein, A., Nakar, D. and Sintov, A., Chondroitin sulphate: A potential biodegradable carrier for colon-specific drug delivery. Int J Pharm, 84: 141-150, 1992.
Rubinstein, A., Nakar, D. and Sintov, A., Colonic drug delivery: Enhanced release of indomethacin from cross linked chondroitin matrix in rat caecal content. Pharm Res, 9: 276-278, 1992.
Sintov, A., Di-Capua, N. and Rubinstein, A., Cross-linked chondroitin sulphate: characterization for drug delivery purposes. Biomaterials, 16: 473-8, 1995.
Tozaki, H., Komoike, J., Tada, C., Maruyama, T., Terabe, A., Suzuki, T., Yamamoto, A. and Muranishi, S., Chitosan capsules for colon-specific drug delivery: improvement of insulin absorption from the rat colon. J Pharm Sci, 86: 1016-21, 1997.
Tozaki, H., Odoriba, T., Okada, N., Fujita, T., Terabe, A., Suzuki, T., Okabe, S., Murnishi, S. and Yamamoto, A., Chitosan capsules for colon-specific drug delivery: enhanced localization of 5-aminosalicylic acid in the large intestine accelerates healing of TNBS-induced colitis in rats. J Control Rel, 82, 51-61, 2002.
Aiedeh, K., and Taha, M.O., Synthesis of chitosan succinate and chitosan phthalate and their evaluation as suggested matrices in orally administered colon-specific drug delivery systems. Arch Pharm (Weinheim), 332: 103-7, 1999.
Tozaki, M., Fujita, T. and Odoriba, T., Colon specific delivery of R68070, a new thromboxane synthetase inhibitor, using chitosan capsules: Therapeutic effects against 2,4,6-trinitrobenzene sulfonic acid-induced ulcerative colitis in rats. Life Sci, 64: 1155-1162, 1999.
Tozaki, M., Emi, Y., Horisaka, E., Fujita, T., Yamamoto, A. and Muranishi, S., Degradation of insulin and calcitonin and their protection by various protease inhibitors in rat caecal contents: Implications in peptide delivery to the colon. J Pharm Pharmacol, 49: 164-168, 1997.
Vandelli, M.A., Leo, E., Forni, F. and Bernatei, M.T., In vitro evaluation of a potential colonic drug delivery system that releases drug after a controllable lag-time. Eur J Pharm Biopharm, 43: 148-151, 1996.
Shu, X.Z., Zhu, K.J. and Song, W., Novel pH-sensitive citrate crosslinked chitosan film for drug controlled release. Int J Pharm, 212: 19-28, 2001.
Zhang, H., Ibrahim, A.A., and Neau, S.H., An in vitro evaluation of a chitosan-containing multiparticulate system for macromolecule delivery to the colon. Int J Pharm, 239: 197-205, 2002.
Shimono, N., Takatori, T., Masumi, T., Ueda, M., Mori, M., Higashi, Y. and Nakamura, Y., Chitosan dispersed system for colon-specific drug delivery. Int J Pharm, 245: 45-54, 2002.
Orienti, I., Cerchiara, T., Luppi, B., Bigucci, F., Zuccari, G. and Zecchi, V., Influence of different chitosan salt on the release of sodium diclofenac in colon-specific delivery. Int J Pharm, 238: 51-59, 2002.
Krogars, K., Heinamaki, J., Vesalahti, J., Marvola, M., Antikainen, O., Yliruusi, J., Marvola, M. and Yliruusi, J., Extrusion-spheronization of pH-sensitive polymeric matrix pellets for possible colonic drug delivery. Int J Pharm, 199: 187-94, 2000.
Nykanen, P., Krogars, K., Sakkinen, M., Heinamaki, J., Jurjensson, H., Veski, P. and Marvola, M., Organic acids as excipients in matrix granules for colon-specific drug delivery. Int J Pharm, 184: 251-61, 1999.
MacNeil, M.E., Rashid, A. and Stevens, H.N.E., Dispensing device. World Patent. WO 9009168, 1990.
g-scintigraphy. Pharm Res, 9: 1436-1441, 1992. Wilding, I.R., Hardy, J.G., Sparrow, R.A., Daviss, S.S., Daly, P.B. and English, J.R., In vitro evaluation of enteric-coated naproxan tablets using
Binns, J.S., Stevens, H.N.E., Bakhashaer, M. and Wilson, G.G., Colon targeted release using the Pulsincap delviery system. Int Symp Controlled Rel Bioact Mater, 21: 260-261, 1994.
Gazzaniga, A., Bussetti, C., Moro, L., Sangali, M.E. and Giordano, F., Time dependent oral delivery system for colon targeting. STP Pharm Sci, 5: 83-88, 1995.
Gazzaniga, A., Iamartino, P., Maffione, G. and Sangalli, M.E., Oral delayed release system for colon specific drug delivery. Int J Pharm, 108: 77-83, 1994.
Gupta, V. K., Beckert, T.E. and Price, J.C., A novel pH- and time-based multi-unit potential colonic drug delivery system. I. Development. Int J Pharm, 213: 1-2:83 - 91, 2001.
Ishibashi, T., Hatano, H., Kobayashi, M., Mizobe, M. and Yoshino, H., Design and evaluation of a new capsule-type dosage form for colon-targeted delivery of drugs. Int J Pharm, 168: 31-40, 1998.
Fukui, E, Miyamura, N., Uemura, K. and Kobayashi, M., Preparation of enteric coated timed release press-coated tablets and evaluation of their function by in vitro and in vivo tests for colon targeting. Int J Pharm, 204: 7 - 15, 2000.
Fukui, E., Miyamura, N. and Kobayashi, M., An in vitro investigation of the suitability of press-coated tablets with hydroxypropylmethylcellulose acetate succinate (HPMCAS) and hydrophobic additives in the outer shell for colon targeting. J Control Rel, 70: 97-107, 2001.
Gupta, V.K., Assmus, M.W., Beckert, T.E. and Price, J.C., A novel pH- and time-based multi-unit potential colonic drug delivery system. II. Optimization of multiple response variables. Int J Pharm, 213: 93-102, 2001.
Pozzi, F., Furlani, P., Gazzaiga, A., Daviss, S.S. and Wilding, I.R., A new oral dosage form for fast and complex release of drug after a predetermined lag time. J Control Rel, 31: 99-104, 1994.
Niwa, K., Takaya, T., Morimoto, T. and Takada, I., Preparation and evaluation of a time-controlled release capsule made of ethyl cellulose for colon delivery of drugs. J Drug Target, 3: 83-89, 1995.
Muraoka, M., Kimura, G., Zhaopeng, H. and Takada, K., Ulcerative colitis--colon delivery of 5-aminosalicylic acid. Nippon Rinsho, 56: 788-794, 1998.
Takaya, T., Niwa, K., Muraoka, M., Ogita, I., Nagai, N., Yano, R., Kimura, G., Yoshikawa, Y., Yoshikawa, H. and Takada, K., Importance of dissolution process on systemic availability of drugs delivered by colon delivery system. J Control Rel, 50: 111-122, 1998.
Muraoka, M., Hu, Z., Shimokawa, T., Sekino, S., Kurogoshi, R., Kuboi, Y., Yoshikawa Y. and Takada, K., Evaluation of intestinal pressure-controlled colon delivery capsule containing caffeine as a model drug in human volunteers. J Control Rel, 52: 119-129, 1998.
Shibata, N., Ohno, T., Shimokawa, T., Hu, Z., Yoshikawa, Y., Koga, K., Murakami, M. and Takada, K., Application of pressure-controlled colon delivery capsules to oral administration of glycyrrizin in dogs. J Pharm Pharmacol, 53: 441-447, 2001.
Sangalli, M.E., Maroni, A., Zema, L., Busetti, C., Giordano, F. and Gazzaniga, A. In vitro and in vivo evaluation of an oral system for time and/or site-specific drug delivery. J Control Rel, 73: 103-110, 2001.
Gingell, R. and Walker, R, Mechanism of azo reduction by non-specificity of Streptococcus faecalis azo reductase. Xenobiotica, 5: 563-571, 1975.
Roxon, J.J., Ryan, A.J. and wright, S.E., Enzymatic reduction of tartrazine by Proteus vulgaris from rats. Food Cosmet Toxicol, 5: 645-656, 1967.
Bragger, J.L., Lloyd, A.W., Soozandehfar, S.H., Bloomfield, S.F., Marriott, C. and Martin, G.P., Investigations into the azo reducing activity of a common colonic microorganism. Int J Pharm, 157: 61-71, 1997.
Larsel, G.L., Larson, J.P. and Gustafsson, J.A., Cystein conjugate beta-lyase in the gastrointestinal bacterium Fusobacterium necrophorum. Xenobiotica, 13: 687-693, 1983.
Schacht, E. and Wilding, I.R., Process for the preparation of azo- and/or disulfide-containing polymers. WO 9111175.
Kimura, Y., Kim, S. and Nishiura, A., Polyurethanes. European Patent. EP 0398472, 1991.
Kimura, Y., Makita, Y., and Kumagai, T., Degradation of azo-containing polyurethane by the action of intestinal flora: its mechanism and application as a drug delivery. Polymer, 33: 5294-5299, 1992.
Gurny, R., Junginger, H.E., Bioadhesion-possibilities and future trends. Wissenchaftliche Verlag, Stuttgart, 1989.
Lenaerts, V. and Gurny, R., Bioadhesive drug delivery systems. CRC Press, Boca Raton, 1990.
Chickering, D.E. and Mathiowitz, E., Bioadhesive microspheres. 1. A novel electrobalance-based method to study adhesive interactions between individual microspheres and intestinal mucosa. J Control Rel, 34: 251-262, 1995.
Chickering, D.E., Jacob, J.S. and Mathiowitz, E., Bioadhesive microspheres. 2. Characterisation and evaluation of bioadhesion involving hard, bioerodible polymers and soft-tissue. Reacting Polymers, 25: 189-206, 1995.
Khosla, R. and Davis, S.S., The effect of polycarbophil on the gastric emptying of pellets. J Pharm Pharmacol, 39: 47-49, 1987.
Haris, D., Fell, J.T., Sharma, H.L. and Taylor, D.C., GI transit of potential bioadhesive formulations in men: A scintigraphy study. J Control Rel, 12: 45-53, 1990.
Kakoulides, E.P., Smart, J.D. and Tsibouklis, J., Azocross-linked poly(acrylic acid) for colonic delivery and adhesion specificity: synthesis and characterisation. J Control Rel, 52: 291-300, 1998.
Kakoulides, E.P., Smart, J.D. and Tsibouklis, J., Azocrosslinked poly(acrylic acid) for colonic delivery and adhesion specificity: in vitro degradation and preliminary ex vivo bioadhesion studies. J Control Rel, 54: 95-109, 1998.
Pellicciari, R. Garzon-Aburbeh and Natalini, B., Brush-border-enzyme-mediated intestine-specific drug delivery. Amino acid prodrugs of 5-aminosalicylic acid. J Med Chem, 36: 4201-4207, 1993.
Rihova, B., Bioadhesive polymers for oral drug delivery. Adv Exp Med Biol, 371B: 1491-1494, 1995.
Brown J.P., McGarrough, G.V., Parkinson, T.M., Wingerd, R.E. Jr. and Onderdonk, A.B., Polymeric drug for treatment of inflammatory bowl disease. J Med Chem, 26: 1300-1307, 1983.
Kopecek, J., The potential of water-soluble polymeric carriers in targeted and site-specific drug delivery. J Control Rel, 11: 279-290, 1990.
Kopecekova, P., Rathi, R., Takada, S., Rihova, B., Berenson, M.M. and Kopecek, J., Bioadhesive N-(2-hydroxypropyl) methacrylamide copolymers for colon-specific drug delivery. J Control Rel, 28: 211-222, 1994.
Jalan, K.N. and Maitra, T.K. Amebiasis in the developing world. In: Ravidin, J.I.(Ed.)., Amebiasis: human infections by Entamoeba histolytica. John Wiley and Sons, New York, pp 535-55, 1988.
Mirelman, D., De Meester, F., Stolarsky, T., Burchard, G.D., Ernst-Cabrera, K. and Wilcheck, M., Effects of covalently bound silica-nitroimidazole drug particles on Entamoeba histolytica. J Inf Dis, 159: 303-309, 1989.
Theeuwes, F., Guittard, G. and Wong, P., Delivery of drugs to colon by oral dosage forms. US Patent 4904474, 1990.
Swanson, D., Barclay, B., Wong, P. and Theeuwes, F., Nifedipine gastro-intestinal therapeutics system. Am J Med, 83 (suppl. 6B): 3, 1987.
Corresponding Author: S.K. Jain, Pharmaceutics Research Projects Laboratory, Department of Pharmaceutical Sciences, Dr. Hari Singh Gour University, Sagar (M.P.), India. drskjainin@yahoo.com
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps