J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(1):67-83, 2003
Development of liposomal polyene antibiotics: an historical perspective.
Agatha W. K. Ng, Kishor M. Wasan1
Division of Pharmaceutics and Biopharmaceutics, Faculty of Pharmaceutical Sciences, The University of British Columbia, Vancouver, British Columbia, CanadaGabriel Lopez-Berestein
Section of Immunobiology and Drug Carriers, Department of Bioimmunotherapy, Division of Medicine, The University of Texas M.D. Anderson Cancer Center, Houston, Texas, USAReceived 14 March 2003, Revised 24 March 2003, Accepted 03 April 2003
PDF version
Abstract
PURPOSE. The purpose of this review article is to review the development of a number of liposomal polyene antibiotics. BACKGROUND: In the past thirty years, the increase in life-threatening pre-systemic and systemic fungal infections within cancer, diabetic and AIDS patients have reached alarming proportions. A number of antifungal agents have been developed to combat this problem. In particular, polyene antibiotics such as Amphotericin B (AmB) and Nystatin (Nys) have remained the most effective and widely used agents in the treatment of these infections. However, their administration is limited by dose-dependent toxicities. One such dose-limiting toxicity is renal toxicity. Polyene antibiotic-induced renal toxicity is believed to be mediated by the drug anchoring to cholesterol within the mammalian cell membrane, resulting in pore formation, abnormal electrolyte flux, decrease in adenosine triphosphate (ATP), and eventually a loss of cell viability. CONCLUSION: In the 1980s and 90s a number of promising lipid-based AmB and Nys formulations were developed to overcome these toxicities. This article will review the development of these liposomal polyene antibiotics.
Introduction
Over the last thirty years, the frequency of life-threatening fungal infections have increased dramatically, particularly among cancer, diabetic and immunocompromised patients.1-4 Several factors have contributed to this rise: improved recognition and diagnosis of fungal infections; prolonged survival of patients with defects in their host defense mechanisms; more invasive surgical procedures; the use of prosthetic devices and indwelling catheters; increased administration of parenteral nutrition; development of resistance fungal strains to currently available drugs; the increase in the number of patients contracting AIDS; and the use of peritoneal dialysis and hemodialysis.5-7 In these patients invasive fungal infections may account for as many as 30% of deaths.8
Polyene antibiotics, Amphotericin B (AmB) (Figure 1A) and Nystatin (Nys) (Figure 1B), derived from fermentation by Streptomyces remain the most effective and widely used compounds in the treatment of both pre-systemic and systemic fungal infections.9,10 However, their administration is limited by dose-dependent toxicities. One such dose-limiting toxicity is renal toxicity. Polyene antibiotic-induced renal toxicity is believed to be mediated by the drug anchoring to cholesterol within the mammalian cell membrane, resulting in pore formation, abnormal electrolyte flux, decrease in adenosine triphosphate (ATP), and eventually a loss of cell viability. Furthermore, these antibiotics interact with ergosterol in the plasma membrane, causing membrane disruption, increased permeability, leakage of vital intracellular constituents, and eventual cell death.11,12 Recent evidence suggests that AmB can cause oxidative damage, which may contribute to its fungicidal activity.13 In general, polyene antibiotics have a higher affinity for the fungal sterol, ergosterol, than its mammalian counterpart, cholesterol, and are thus less toxic to mammalian cells.14
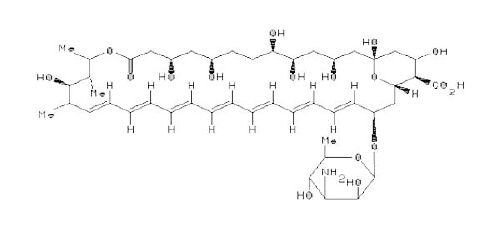
Figure 1: A Chemical Structure of Amphotericin B (AmB)
Reference: 10. http://chem.sis.nlm.nih.gov/aidsdrg4.html
Figure 1: B Chemical Structure of Nystatin (Nys)
Reference: 10. http://chem.sis.nlm.nih.gov/aidsdrg4.html
Since the clinical use of these compounds has been limited by their toxic effects,15-17 an important question is how to best direct the drug specifically to the fungus and keep it away from sites of toxicity. One strategy is to incorporate these polyene antibiotics into lipid-based delivery systems to enhance their therapeutic index by decreasing their rate-limiting toxicity and by increasing target specificity. This paper will review the development and current use of a number of liposomal polyene antibiotics.
Development Of Liposomal Amphotericin B (L-Amb) And Amphotericin B Lipid Complex (Ablc)
Background
In the early 1980s, several research groups developed new AmB formulations by incorporating the drug into liposomes. New and coworkers initially investigated the antileshmanial activity of AmB and other antifungal agents entrapped in liposomes.18 Six months later, Graybill and coworkers published the first extensive paper investigating the treatment of murine cryptococcosis with liposome-associated AmB.19 In this study, BALB/c mice were challenged with Cryptococcus neoformans and given liposome-associated AmB or AmB-deoxycholate intravenously. They found mice that were treated with liposome-associated AmB survived longer and had lower tissue counts of cryptococci than mice treated with AmB-deoxycholate or untreated control mice. They concluded that the reduced acute toxicity of liposome-associated AmB permitted much larger doses of AmB to be given than were possible with AmB-deoxycholate.
In the early 1980s, our laboratory incorporated AmB into liposomes consisting of dimyristoyl phosphatidylcholine (DMPC) and dimyristoyl phosphatidylglycerol (DMPG) in a lipid-to-drug weight ratio of 12:1 to form liposomal Amphotericin B (L-AmB).20-27 We have shown, in both experimental and clinical studies that this formulation has less toxicity than conventional AmB, which allows increased doses to be given.
Pre-Clinical Studies
The effectiveness of free AmB (Fungizone®, Bristol-Myers Squibb, Nutley, NJ, USA; consisting of AmB and sodium deoxycholate) and that of L-AmB were tested against experimentally induced systemic candidiasis in mice.20 Mice were inoculated intravenously with a strain of Candida albicans isolated from a patient with systemic candidiasis. Two days after the inoculation, a severe infection was detected in the liver, spleen, and kidneys. All treatments were administered intravenously starting 2 days after the inoculation with Candida albicans. Empty liposomes did not affect mouse survival. Multiple doses of free AmB at its maximum tolerated dose enhanced survival of mice significantly; however, a similar regimen using L-AmB was superior to one with free AmB and led to prolonged survival (greater than 60 days) and to a 60% cure rate (no histopathologic or microbiologic evidence of infection). L-AmB injected as a single cumulative dose corresponding to the total dose of the other regimens led to a statistically significant enhancement in survival compared with the result obtained with free AmB. These data demonstrate that L-AmB is far more active than free AmB in this model. The findings in experimentally induced candidiasis were later confirmed by others.29,30 AmB incorporated into liposomes was also shown to be effective against experimentally induced histoplasmosis31 and induced leishmaniasis in hamsters and nonhuman primates.32 L-AmB was shown to be from 331 to 750 times more active than meglumine antimonate and from 2 to 5 times more active than free AmB in hamsters infected with Leishmania donovani. In squirrel monkeys infected with Leishmania donovani, L-AmB led to a 99% suppression of amastigotes in the liver. In two murine models of cutaneous leishmaniasis, no significant decrease in tissue parasite density was observed when treated with liposome-intercalated AmB seven days following inoculation with Leishmania tropica.33 However, a case of visceral leishmaniasis in mice unresponsive to several courses of treatment with standard antifungal agents, was successfully cured by a 21-day course (50 mg/day) of L-AmB (AmBisome, Vestar, Inc.). 34 Furthermore, AmBisome® appears to be effective following multiple dose-therapies to mice infected with Leishmania infantum.35
These experiments with L-AmB suggest that the therapeutic index is increased due to the better tolerance of high AmB dosages, but that the efficacy of a given dose is similar or even slightly decreased with L-AmB. However, Leishmaniasis appears to be an exception since L-AmB efficacy is increased at low unitary dosage.
Clinical Studies
Early clinical trials with L-AmB within patients with systemic mycoses refractory to free AmB and other antifungal agents were conducted at The University of Texas M. D. Anderson Cancer Center, Houston, Texas in the 1980s.27,36,37 Though the dosage was modified according to each patient's tolerance, the standard regimen was 2 mg/kg body weight of AmB daily for 3 days, which, when well tolerated (no fever, chills, or changes in kidney function), was increased by 1 mg/kg every fourth dose until 5 mg/kg was reached. Then 5 mg/kg of AmB was infused once daily for 3 days until the patient had received a total of 75-mg AmB/kg. The maximal single dose administered in those studies was 6 mg/kg.
All patients tolerated L-AmB well; mild fever and chills occurred in only a few. Potassium supplements were required in most patients, particularly in those who received doses greater than 2-mg/kg-body weight of L-AmB. Clinical improvement was observed in most patients during the first week of treatment, and no long-term renal, hepatic, or central nervous system toxicities were observed.
An additional study included 46 cancer patients who had developed a variety of systemic fungal infections and were treated with L-AmB.37 Twenty-one of these patients had disseminated candidiasis, 19 had aspergillosis, and the rest had a variety of other fungal infections. Forty patients failed to respond to conventional AmB therapy, and 6 were given L-AmB because therapy with conventional AmB drugs had caused severe side effects (e.g., nausea, vomiting, and hypokalemia). Twenty-four patients had a complete response, and 22 patients had none. No short- or long-term toxicities were observed. Acute side effects associated with conventional AmB therapy (e.g., fever, chills, and potassium loss) were infrequent and milder in patients given L-AmB than commonly observed in patients given conventional AmB. No chronic renal, hematological, or central nervous system side effects were observed following therapy with L-AmB.
L-AmB is effective and less toxic than free AmB in the treatment of fungal infections caused by Candida albicans and Aspergillus niger; even in patients with neutropenia. The administration of L-AmB allowed for antileukemic treatment despite the presence of an active fungal infection and chemotherapy-induced neutropenia, which usually compromises treatment of the fungal infection. L-AmB therapy is easier than AmB therapy, in part because of lower fluid volumes and shorter intravenous infusion times that enable patients to continue antifungal treatment as outpatients.
Furthermore, it has been observed two cases of visceral leishmaniasis within patients infected with HIV that L-AmB has been effective in curing these patients without toxicity.38 Fusai and coworkers have also reported L-AmB to be an effective anti-leishmania agent resulting in complete remission of polyresistant visceral leishmaniasis following L-AmB therapy.39 L-AmB has been reformulated and marketed as Amphotericin B Lipid Complex (ABLC, Abelcet®, The Liposome Company Inc., Princeton, NJ, USA). Recently, a number of studies evaluating the efficacy, safety, and pharmacokinetics of ABLC in different patient populations have been published. Linden et al.40 reported the efficacy and safety of ABLC administration in solid-organ transplant recipients with invasive fungal infections. They found that within 79 solid-organ transplant recipients who were refractory to or intolerant to conventional antifungal therapy (mainly AmB) or had pre-existing renal disease ABLC therapy to be effective (clinical response rate of 58%) and safe (renal-sparing properties). Adedoyin et al.41 reported a pharmacokinetic evaluation of ABLC within patients with definite or probable systemic fungal infections following daily intravenous infusions for 10 to 17 days. They observed that AmB exhibited a multicompartment plasma concentration versus time profile with high systemic clearance, a large volume of distribution at steady state, and a long terminal half-life. However, no evidence of AmB accumulation within the systemic circulation after multiple daily doses was observed. These investigators suggest that ABLC may exist as a depot in the tissues from which free AmB is slowly released to limit systemic exposure.
Mechanistic Studies
Phagocyte Transport of L-AmB:
Phagocytes in the circulation and in tissues avidly take up liposomes. Others42-44 and we have shown that liposomes are distributed in animals and man in organs rich in mononuclear phagocyte system cells. We previously observed that liposome incorporation enhanced the delivery of AmB to Candida infected organs in mice.24 A potential exists, therefore, that monocytes and macrophages in peripheral blood may take up the drug-laden liposomes and transport them to the infected sites.
The in vitro uptake of AmB and L-AmB by murine peritoneal macrophages was studied.45 Resident peritoneal macrophages were incubated at several time intervals with L-AmB or AmB in RPMI 1640 supplemented with 10% fetal calf serum. After each time interval, the supernatants were discarded, and the monolayer was washed three times with warm phosphate-buffered saline. The cells were then lysed, and radioactivity in the cell lysates was measured. Maximal uptake of L-AmB was observed after 8 hours, with a gradual decrease from 24 to 72 hours. The macrophage's liposome uptake capacity was maximal at 8 hours with a lipid saturation capacity of 100 mg lipid per million macrophages. No uptake was observed for AmB. However, recovery of the drug in organs rich in mononuclear phagocytes does not necessarily mean that phagocytes take up the drug in vivo.
In the setting of patients with neutropenia, phagocytic transport is less likely to play a major role. Furthermore, since resident macrophages are less sensitive to cytotoxic agents than are other cell types, we believe that in the neutropenic setting a different transport of AmB may exist.
Lipoprotein Transport of L-AmB and ABLC:
Serum lipoproteins have been hypothesized to influence the pharmacokinetics, tissue distribution, and pharmacological activity of L-AmB in rats.46 In man, large volume of distribution of AmB appears to be a result of the drug's high accumulation in the kidney, liver, and lung tissues.47 Injection of drug-free liposomes (DMPC:DMPG 7:3 w/w ratio) into the human circulation has resulted in a large volume of distribution and a long terminal half-life.48 When AmB was injected intravenously into mice, only 15% of the original dose could be accounted for, 10% in the lung and 5% in the liver.49 Furthermore, pharmacokinetics studies in humans have shown AmB to have a long terminal half-life (t1/2b= 15 days), and a very short distribution half-life (t1/2a).47 Intravenous injection of AmB into animals has resulted in slow or sustained release of the drug and altered tissue kinetics and distribution.50 It has been suggested that the unusual pharmacokinetics of AmB may be a result of the slow release of the drug from a tissue or organ site due to the high-affinity binding of it to cholesterol in serum lipoproteins or cell membranes.51-54
Brajtburg and coworkers examined the interactions of AmB with human serum lipoproteins in vitro in an attempt to understand these interactions and how they might affect the pharmacological behavior of AmB. Their studies showed AmB to be equally associated with high-density lipoprotein (HDL) and low-density lipoprotein (LDL) fractions after 1 hour of incubation at 25°C.51 Furthermore, AmB injected in LDL to rabbits was toxic: 70% of the rabbits died from a non-toxic dose of AmB (1.0 mg/kg), which implies that LDL association would increase AmB toxicity.53
The results we obtained demonstrated that changes in temperature and association with lipid-complexes affect the distribution of AmB in serum lipoproteins.55 Similar to the results reported by Brajtburg et al.,53 when serum lipoproteins were separated by affinity chromatography, an equal distribution of AmB was found in human serum lipoprotein fractions following one-hour incubation at 25°C. In contrast, at 37°C, over 90% of the concentration of AmB was found in the HDL fraction following 1 hour incubation. However, recent studies in our lab using density gradient ultracentrifugation to separate plasma lipoproteins suggest this is an overestimation of the percentage of AmB recovered in the HDL fraction.56 In this study, we found between 66-80% of the original concentration incubated in plasma was recovered in the lipoprotein-deficient plasma fraction, which contains mainly albumin and a-1-glycoprotein. This inconsistency in results is due to the limitation of the affinity chromatography technique in determining the lipoprotein distribution of drugs. This technique can only separate LDL and VLDL away from the other plasma proteins and lipoproteins. It cannot separate HDL from the plasma proteins (albumin and a-1-glycoprotein). However, using density-gradient ultracentrifugation, the different lipoproteins can be separated away from plasma proteins.
Studies were subsequently conducted where human serum was incubated with ABLC (DMPC:DMPG 7:3 w/w ratio with a DMPG:AmB 4:1 M ratio) for 60 minutes at 37°C, serum was separated into its lipoprotein fractions by affinity chromatography, and DMPG and AmB was quantified by HPLC. Ninety percent of the drug and 80% of the lipid was found in the HDL fraction in a 3:1 M ratio (DMPG: AmB), while in a 6:1 M ratio (DMPG:AmB) in the LDL fraction. When these studies were repeated whereby serum lipoproteins were separated by density-gradient ultracentrifugation, 60-90% of the original concentration of AmB (as ABLC) incubated was recovered in the HDL (Figure 2).56 Taken together, these experiments further suggested that AmB and DMPG may co-transfer as a intact drug-lipid complex to serum lipoproteins.
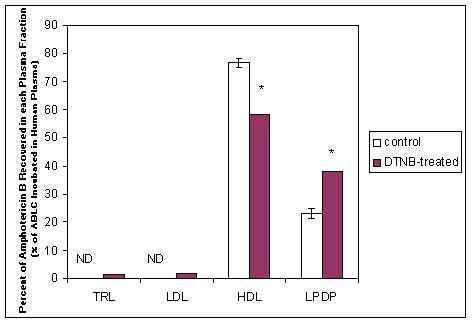
Figure 2: Distribution of Amphotericin B Lipid Complex (ABLC) (20μg of AmB/ml of plasma) following incubation within human plasma that has been treated with DTNBa (15mM). ABLC was incubated in plasma pretreated with DTNBa 18 hours prior to the experiment or in untreated plasma (control) for 60 min at 37oC. Following incubation the plasma was separated into its triglyceride-rich lipoprotein (TRL), low-density lipoprotein (LDL), high-density lipoprotein (HDL), and lipoprotein-deficient plasma (LPDP) fractions by ultracentrifugation and assayed for AmB by HPLC; n = 3; data are expressed as mean ± standard deviation. * P < 0.05 vs. control.
DTNB (dithionitrobenzoate, 15mM, a compound that inhibits lecithin:cholesterol acyltransferase conversion of HDL3 free cholesterol to esterified cholesterol, resulting in an elevation of HDL3 coat lipid concentrations. This finding suggests that alterations in HDL lipid composition influence the plasma distribution of ABLC.).
Reference: 56. A. L. Kennedy and K. M. Wasan, J. Pharm. Sci. 88, 1149-55 (1999).
The modification of the distribution of AmB to serum lipoproteins at 37°C following ABLC incubation may be related to the transition temperature of lipoproteins, which is between 27°C and 34°C.57,58 At the transition temperature, cholesteryl esters within the lipoprotein core exist as an isotropic solution, while below this temperature, they form disordered smectic liquid crystals.57,58 The core of HDL becomes more ordered at the higher temperature, thus making it easier for the AmB molecule to associate with it. This hypothesis is based on the assumption that AmB is incorporated into the lipophilic core of these lipoproteins.
DMPG as an anionic exogenous phospholipid may distribute into HDL as opposed to LDL and be partially responsible for the concurrent transport of AmB to HDL. Since HDL and LDL are not found in an equimolar ratio in human serum, but at an LDL:HDL ratio of 6:1,55 the data suggest that some mechanism besides random collision must drive this drug-liposome complex towards HDL rather than LDL. When ABLC (4:1 molar ratio DMPG:AmB) was incubated for 1 hour at 37°C in human serum, AmB and DMPG seemed to co-transfer to the serum lipoproteins.55 These observations suggest that phospholipids with a negative charge may be responsible for the altered AmB-lipoprotein distribution patterns. In addition, work by Surewicz and coworkers has suggested the formation of thermally stable complexes between anionic phospholipids such as DMPG and apolipoprotein AI, one of the predominant protein components associated with HDL.59 Furthermore, the DMPG: AmB mole ratio found in the lipoprotein fractions is similar to the initial mole ratio of the liposomes prior to incubation, which suggests that the drug-lipid association remains intact as it travels to HDL.
Pharmacological implications of the AmB-lipoprotein complex
Preliminary investigations by others have suggested that the renal toxicity of AmB can be influenced by liposomal-phospholipid surface charge, phospholipid acyl chain length, chain saturation, and the liposomal-lipid/AmB ratio.60,61 For example, AmB-containing liposomes composed of phospholipids with unsaturated acyl chains are as toxic as AmB to mammalian cells; however, those composed of phospholipids with saturated acyl chains are less toxic.60,62 Previous studies have demonstrated a decrease of AmB cytotoxicity when the drug is delivered in the form of L-AmB to LLC PK1 cells (a pig kidney epithelial cell line)60,63 and to primary cultures of rabbit proximal tubule cells.64 To date, the mechanisms that result in the decreased renal cytotoxic effects of L-AmB are not fully understood. Krause and Juliano have suggested that the decreased toxicity of L-AmB compared with AmB is related to a selective transfer of the drug from liposomes to fungal but not mammalian cell membranes.63 This selective toxicity shown towards the fungal membrane is probably regulated by physical characteristics of the donor and of the target membrane.60 Brajtburg and coworkers demonstrated that AmB is highly bound to plasma lipoproteins51 and that AmB-induced cytotoxic effects on mammalian red blood cells, but not Candida albicans cells decreased in the presence of either HDL or LDL.65 Furthermore, Barwicz and coworkers have suggested that AmB association with LDL and VLDL may be responsible for AmB nephrotoxicity in vivo and that hindering this complex formation result in a decrease in AmB nephrotoxicity.66
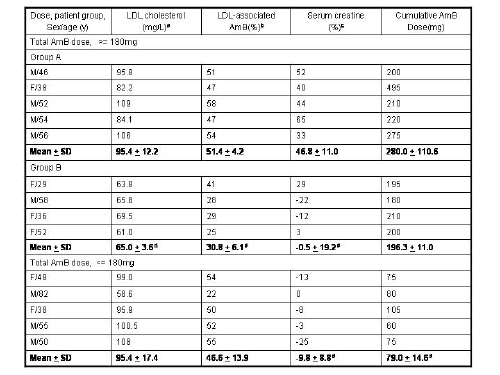
Previously, we demonstrated that HDL-associated AmB and HDL-associated L-AmB are less toxic to LLC PK1 renal cells than are AmB or LDL-associated AmB (Figure 3).67 The reduced toxicity of HDL-associated AmB may be explained by the low level of expression of HDL receptors in LLC PK1 cells. The sustained toxicity observed with AmB alone in trypsinized cells may be related to a direct membrane effect. However, when AmB is associated with LDL, the toxicity is maintained, which suggests that both direct membrane- and non-membrane-related toxicity may occur. Furthermore, a study to determine if a relationship existed between serum lipoprotein cholesterol concentration and the severity of AmB-induced renal toxicity in patients suggested that patients with higher serum LDL-cholesterol concentrations are more susceptible to AmB-induced renal toxicity (Table I).66
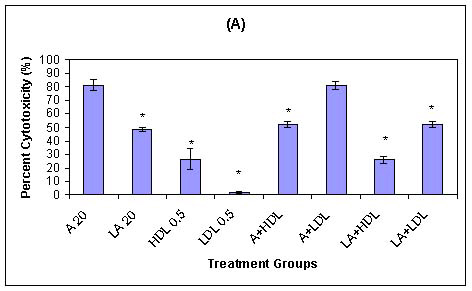
Figure 3: (A) Influence of serum Lipoprotein-associated AmB and L-AmB on drug toxicity to LLC PK1 renal cells as percent cytotoxicity of LLC PK1 cells in serum-free medium at 37oC containing the following treatments: AmB (20 mg/ml) (A 20), L-AmB (20 mg/ml) (LA 20), HDLs (0.5mg of protein per ml; HDL 0.5), LDLs (0.5 mg of protein per ml; LDL 0.5), mixtures of AmB (20 mg/ml) with HDLs (A+HDL) or LDLs (A+LDL), and mixtures of L-AmB (20 mg/ml) with HDLs (LA+HDL) or LDLs (LA+LDL) (n = 3; values are means ± standard deviations). * P < 0.05 versus treatment with AmB at 20 mg/ml.
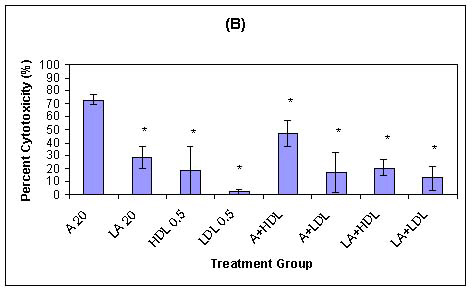
Figure 3: (B) Influence of reduced LDL receptor expression on HDL- and LDL-associated AmB toxicity of LLC PK1 renal cells as percent cytotoxicity of LLC PK1 cells in serum-free medium at 37oC after a trypsin wash containing the same treatments as described for panel A (n = 3; values are means ± standard deviations). * P < 0.05 versus treatment with AmB at 20 mg/ml.
Reference: 67. K. M. Wasan, M. G. Rosenblum, L. Cheung and G. Lopez-Berestein, Antimicrob. Agents Chemother. 38, 223-7 (1994).
Table 1: The serum low-density lipoprotein (LDL) cholesterol concentration, amount of AmB associated with plasma LDL, serum creatinine concentrations, and cumulative AmB dose following 10 days of therapy administered to patients with an anticipated or confirmed fungal infection.
Reference: 68. K. M. Wasan and J. S. Conklin, Clin. Infect. Dis. 24, 78-80 (1997).
HDL and LDL associated-AmB were equally toxic to fungal cells, which suggest that the presence of lipoproteins does not alter the antifungal activity of AmB. Such effects may be related to the liberation of monomeric AmB associated with lipoproteins or L-AmB, by fungal or endothelial derived phospholipases.61,62 The low concentrations of unbound and water-soluble monomeric AmB present in L-AmB69-71 may be sufficient for fungal toxicity but not adequate for forming AmB aggregates that are toxic to mammalian cells.62,63 AmB complexed with lipid is less toxic than the self associated form of AmB in medium, but the monomeric form of AmB interacts with fungal cells membrane and is non-toxic against mammalian cells membrane as shown by Bolard et al.70
Differences in the pharmacokinetics and tissue distribution of free AmB were demonstrated in healthy in comparison to hyperlipidemic rats induced with diabetes. In contrast, the pharmacokinetics and tissue distribution of L-AmB were unchanged in diabetic rats which suggests an independence of this delivery mechanism from the diabetic disease state and endogenous triglyceride and cholesterol levels.46 However, a limitation of this study was that we could not determine if changes in the pharmacokinetics and tissue distribution of AmB was a direct result of the plasma hyperlipidemia or other diabetic-inflicted physiologic alterations (e.g. blood flow, liver metabolism, renal metabolism). Wasan and Conklin suggest that following administration of a single intravenous dose, AmB and L-AmB appear to be less effective in killing Candida albicans isolates in hypercholesterolemic diabetic than in normocholesterolemic nondiabetic rats, while they were found to improve the renal functions of rats in both treatment groups.72
An explanation for these findings maybe due to changes in the pharmacokinetics and tissue distribution of AmB and L-AmB in hypercholesterolemic blood.
To determine if the pharmacokinetics and tissue distribution of AmB and L-AmB were altered in plasma dyslipidemia (hypercholesterolemia) independent of other physiologic alterations, rats were administered a continuous infusion of Intralipid®. Intralipid® is a fatty acid/triglyceride emulsion administered intravenously as a nutritional supplement in debilitated patients. We found that in rats administered a continuous infusion of Intralipid® for 5 days resulted in an increase in total serum cholesterol and HDL cholesterol concentrations without altering LDL cholesterol or total serum triglyceride concentrations.73
The influence of 5% Intralipid, and 0.45% normal-saline infusions on the concentration in serum and distribution in tissue of AmB (Fungizone, consisting of AmB and sodium deoxycholate) and L-AmB in rats were compared.74 In animals receiving a continuous Intralipid, infusion, concentrations of AmB in kidneys and lungs were significantly higher, but the concentration of AmB in serum was significantly lower in animals administered AmB versus those given L-AmB. In animals receiving a continuous normal-saline infusion concentrations of AmB in kidneys and the spleen were significantly higher, but the concentration of AmB in serum was significantly lower in animals administered AmB versus those given L-AmB. These results suggest that the increased total serum cholesterol and high-density lipoprotein cholesterol during the Intralipid, infusion decreased the clearance of AmB from the bloodstream and decreased the L-AmB concentration in the kidney and lung.
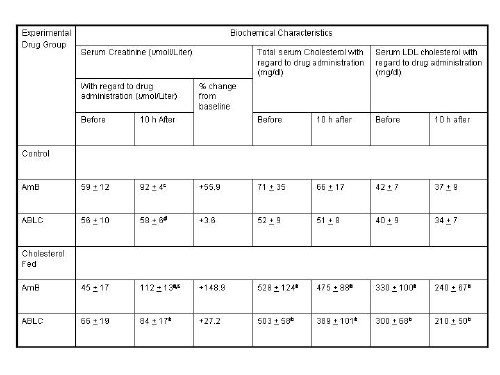
Wasan and coworkers have recently investigated the pharmacokinetics, tissue and plasma distribution and renal toxicities of AmB following single- and multiple-dose administration to rabbits with elevated plasma cholesterol levels.75,76 Following 7 days of administration of a cholesterol-enriched diet (0.5% w/v) or regular rabbit diet, each rabbit was administered a single intravenous bolus of AmB or ABLC (1.0 mg/kg) daily for 7 consecutive days. Before drug treatment, cholesterol-fed rabbits demonstrated marked increases in total plasma LDL and triglyceride-rich lipoprotein cholesterol levels compared with levels in rabbits on regular diet. No differences in total plasma triglyceride levels were observed. Significant increases in plasma creatinine (as a marker of renal toxicity) were observed following AmB administration to cholesterol-fed and regular-fed rabbits (Table II). However, the magnitude of this increase was two-fold greater in rabbits fed a regular diet than in rabbits fed a cholesterol-enriched diet. An increase in plasma creatinine levels was observed only in rabbits on a cholesterol-enriched diet administered ABLC (Table II). The pharmacokinetics of AmB was significantly altered in rabbits on a cholesterol-enriched diet administered AmB or ABLC compared to those rabbits on a regular diet (Table II). An increased percentage of AmB was recovered in the triglyceride-rich lipoprotein fraction when AmB was administered to rabbits fed a cholesterol-enriched diet than when it was administered to rabbits fed a regular diet (Figure 4). Furthermore, an increased percentage of AmB was recovered in the LDL and triglyceride-rich lipoprotein fractions when ABLC was administered to rabbits fed a cholesterol-enriched diet than when fed a regular rabbit diet (Figure 4). Taken together, these findings suggest that an increase in plasma cholesterol levels modifies the pharmacokinetics of AmB and renal toxicity following administration of multiple intravenous doses of AmB and ABLC.
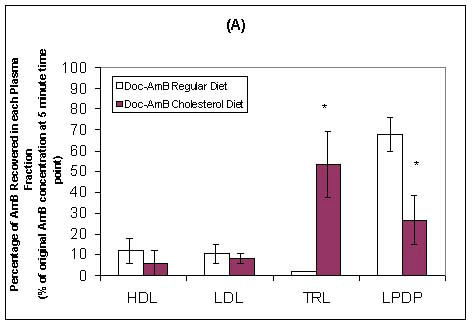
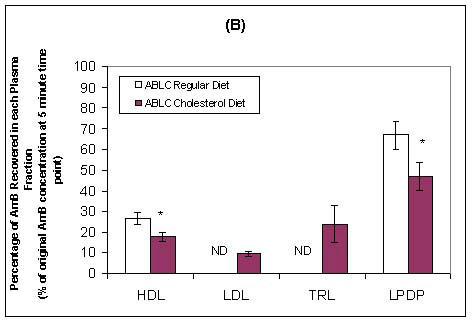
Figure 4: In vivo distribution in plasma at 5 minutes following the administration of the last intravenous dose of Doc-AmB (A) or ABLC (B) to cholesterol-fed or regular diet-fed rabbits. Values are means ± standard deviations (n = 5), * P < 0.05 versus rabbits fed a regular diet and receiving Doc-AmB or ABLC. The LPDP fraction includes albumin and α-1-glycoprotein.
Reference: 76. M. Ramaswamy, K. D. Peteherych, A. L. Kennedy and K. M. Wasan, Antimicrob. Agents Chemother. 45, 1184-91 (2001)
Table 2: Biochemical Characteristics of serum and pharmacokinetic parameters of drug after a single intravenous dose of AmB and ABLC (1mg/kg) in control and cholesterol-fed (0.05% w/v cholesterol) rabbits.a
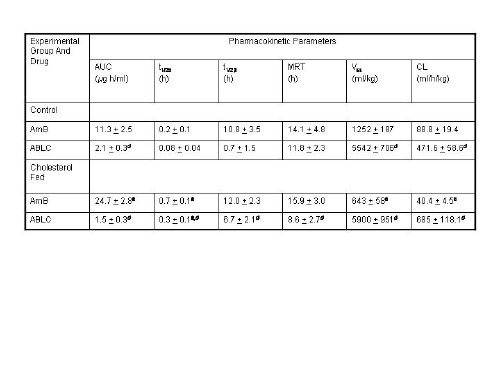
Reference: 75. K. M. Wasan, A. L. Kennedy, S. M. Cassidy, M. Ramaswamy, L. Holtorf, J. W. Chou and P. H. Pritchard, Antimicrob. Agents Chemother. 42, 3146-52 (1998).
New Applications For Amphotericin B Lipid Complex (Ablc)
Recently a number of clinical and pre-clinical studies describing new applications of ABLC have been published. Palmer and coworkers reported the safety of aerosolized ABLC in lung transplant recipients.77 They found aerosolized ABLC was well tolerated within 98% of patients (51 in total) administered the formulation. Pulmonary mechanics worsened by 20% or more in less than 5% of patients post treatment. Furthermore, there were no significant adverse effects related to study medication in any patient and 1-year survival for all enrolled patients were 78%.
Others have reported the use of ABLC in pediatric and neonatal populations. Knoppert et al.78 reported the eradication of severe neonatal systemic candidiasis in a preterm female infant, born at 25 weeks' gestational age with a birth weight of 870 g with ABLC. At seven weeks of age, this child developed severe disseminated candidiasis, which failed to respond to conventional AmB and fluconazole therapy. Her progressive deterioration was reversed only after ABLC was substituted for conventional AmB and she made a full recovery without any adverse effect. Herbrecht et al.79 reported on the efficacy of ABLC in the treatment of invasive fungal infections in immunosuppressed pediatric patients. This study included a large proportion of patients who were refractory to or intolerant of conventional antifungal therapy with a primary underlying condition including haematopioetic stem cell transplantation, leukaemia and lung transplantation. Following daily therapy for 38 days at a mean dose of 4 mg AmB/kg/day, 83% of the patients responded successfully to the ABLC treatment with low incidence of adverse events. Taken together, these studies support the use of ABLC in the treatment of invasive fungal infections in children, including patients who have previously failed, or are intolerant of traditional antifungal regimens.
In addition, recent cases have been published utilizing ABLC in unusual circumstances. Goldblum et al.80 reported the first case of Fusarium solani keratitis that progressed to fungal endophthalmitis and was successfully treated with ABLC. In this case, a 34-year-old immunocompromised woman developed a contact lens-related Fusarium solani keratitis. Subsequently this patient developed fungal endophthalmitis, which was eventually treated with ABLC after systemic AmB and ketoconazole in addition to topical natamycin and AmB proved ineffective. Cook reported the first case of cerebral blastomycosis successfully treated with ABLC in a patient previously treated with itraconazole.81
Development Of Other Lipid-Based Amphotericin B Products
Two other AmB lipid-based formulations have also been prepared on a large scale and are available for clinical use. AmB colloidal dispersion (ABCD; Amphocil,; SEQUUS Pharmaceuticals, Menlo Park, CA, USA) is a stable complex of AmB and cholesteryl sulfate in a 1:1 molar ratio.82 ABCD has equivalent antifungal activity but decreased toxicity than does the commercially available form of AmB, AmB plus deoxycholate (Fungizone,, Bristol-Myers Squibb, Nutley, NJ, USA).82 In vitro studies have shown that the drug-lipid complex does not hemolyze erythrocytes and binds less to plasma lipoproteins than does the conventional form of AmB.83,84 Studies in healthy volunteers indicated that drug disposition of ABCD was similar to that of Fungizone,, Acute side effects after ABCD administration were comparable with those of AmB but occurred at doses with 1.5 mg/kg/day compared to 0.5-0.75 mg/kg/day with the conventional preparation. The renal toxicity of ABCD is believed to be reduced because the AmB is bound, as a cholesterol complex, so less "free" drug is available to interact with renal tubules.84
AmBisome® (Vestar, San Dimas, CA, USA)83,85 is supplied as a lyophilized powder, which must be reconstituted before intravenous infusion. It is the only liposomal AmB preparation currently licensed in the United Kingdom. This formulation consists of hydrogenated soy phosphatidylcholine, cholesterol, distearoyl phosphatidylglycerol, a-tocopherol, sucrose, and disodium succinate hexahydrate. A starting dose of 1.0 mg/kg/day has been recommended, increasing to 3.0 mg/kg/day, although doses up to 5.0 mg/kg/day have been used in compassionate studies, where exposure to the conventional AmB preparation led to unacceptable toxicity.85-87 The highest concentrations of AmBisome® are found in the liver and spleen; however, concentrations in the lung and kidney are highly inconsistent. A recent study by Fleming et al.88 compared AmBisome® to ABLC in the treatment of suspected or documented fungal infections in patients with leukemia. They reported that ABLC and AmBisome® were equally effective for the treatment of suspected or documented fungal infections. Although AmBisome® was better tolerated than ABLC when administered to patients in this study, AmBisome® administration was still associated with mild abnormalities in liver function at the end of therapy.
Development Of Liposomal Nystatin (L-Nys)
Background
Nystatin (Nys) is a polyene antifungal antibiotic that has been used for the treatment of cutaneous, vaginal, and oral fungal infections since the 1950s.89.90 Topical and oral administration of Nys are not associated with significant toxicities because the compound is not absorbed through the skin or from the gastrointestinal tract.91 To overcome its limited bioavailability Nys has been administered intravenously. Previous attempts to administer Nys parenterally in the clinic resulted in sclerosing of the veins accompanied by severe shaking, chills, fever and malaise. This eventually resulted in the discontinuation of Nys therapy. To circumvent the problems associated with parenteral administration of free Nys, Aronex Pharmaceuticals Inc. has recently developed Nyotran,, a liposomal formulation of Nys.
Development of Nyotran, for intravenous administration provides an additional compound with which the physician would be able to treat systemic fungal infections. Although numerous studies demonstrate that while the fungicidal activities of Nys and AmB overlap, they are not identical. For example, Candida albicans strains exist that are markedly more sensitive to Nys than to AmB.92 Nys was found to be effective against Geotrichum, Torulopsis, Candida krusei, and Beauvaria, whereas AmB has no activity against these fungi. To date a number of studies investigating the toxicity and efficacy of Nyotran, have been published.
Pre-Clinical Studies
Our group was one of the first to investigate the in vitro and in vivo toxicity and therapeutic effects of liposomal Nys.93,94 Nys incorporated into multilamellar lipid vesicles was as active as unincorporated Nys was against a wide variety of yeasts and fungi, including Candida albicans. In addition, Nys incorporation into these liposomes also protected erythrocytes from the toxic effects of unincorporated Nys. Further studies were completed in mice infected with Candida albicans. Mice injected intravenously with various doses of free and liposomal Nys showed a significant decrease in Nys-induced toxicity after Nys was incorporated into the liposomes. The maximum tolerated does of mice treated with free Nys was 4 mg/kg while those treated with liposomal Nys had a maximum tolerated dose of 16 mg/kg. The maximal tolerated dose of free Nys had no effect in the treatment of mice infected with Candida albicans, whereas mice administered an equivalent dose of liposomal Nys had greater survival rates. Take together, these studies suggested that incorporation of Nys into liposomes reduced its toxicity without compromising its antifungal activity.
Over the past several years (1997-2001) the efficacy, safety and pharmacokinetics of liposomal Nys in a variety of different animal models has been reported. Wallace et al.95 reported the activity of liposomal Nys against disseminated Aspergillus fumigatus infection in neutropenic mice. They found liposomal Nys, at doses as low as 2 mg/kg/day protected neutropenic mice against Aspergillus-induced death to a statistically greater extent than the no-treatment, saline control, or the empty-liposome group. Similar protection was observed when these mice were administered free Nys. In addition, histopathological results demonstrated that following only 3 doses of liposomal Nys the lungs, spleen, pancreas, kidney and liver were clear of Aspergillus. Groll et al.96,97 reported two studies investigating the safety and efficacy of liposomal Nys in neutropenic rabbits that had either pulmonary aspergillosis or subacute disseminated candidasis. They found liposomal Nys increased survival, reduced tissue injury, was effective in clearing the infection and had tolerable side effects when administered to neutropenic rabbits with pulmonary aspergillosis. They further found liposomal Nys less renal toxic than AmB and had dose-dependent activity comparable to that of AmB for the early treatment of subacute disseminated candidiasis in these rabbits. Groll and coworkers have also reported on the pharmacokinetics and tissue distribution of liposomal Nys following single and multiple daily intravenous administration to rabbits. They observed that liposomal Nys displayed nonlinear pharmacokinetics, potentially therapeutic peak plasma concentrations, and substantial penetration into tissues.98
In the last several years, a number of studies have been published evaluating the safety and effectiveness of liposomal Nys compared to other antifungal agents including AmB, liposomal AmB, itraconazole and fluconazole. Oakley and coworkers compared the in vitro activity of liposomal Nys with those of other antifungal agents against 60 Aspergillus isolates. Twelve isolates were resistant to itraconazole. They found liposomal Nys to have a lower MIC than Nys and AmB lipid complex, but a higher MIC than itraconazole (nonresistant strains), AmB, liposomal AmB and AmB colloidal dispersion.99 Denning et al.100 compared different doses of liposomal Nys, liposomal AmB, ABLC with AmB alone in neutropenic mice infected with an isolate of Aspergillus fumigatus. They found that high doses of liposomal AmB and ABLC (25 and 50 mg/kg) could overcome reduced susceptibility to AmB, but all were inferior to 5- and 10-fold lower doses of liposomal Nys. Carrillo-Munoz et al.101 compared the in vitro susceptibility of 120 clinical isolates of yeasts to liposomal Nys with that of ABLC, liposomal AmB, AmB cholesteryl sulphate, AmB, Nys, fluconazole and itraconazole. No significant differences in MICs were seen between the activity of liposomal Nys and the polyene drugs or itraconazole, but liposomal Nys was more active than fluconazole. Taken together, these studies indicate that liposomal Nys has good activity in vitro against medically important yeasts, and compares favorably with the activities of other currently available antifungal agents, but with apparently less systemic toxicity.
Our laboratory has recently investigated the plasma distribution of free and liposomal Nys within human, dog and rat plasma.102 When free and liposomal Nys were incubated in human, dog or rat plasma, the majority of the drug was recovered in the lipoprotein-deficient plasma fraction (which primarily contains albumin and a-1-glycoprotein) (Figure 5). However, a significantly greater percentage of Nys was recovered in the lipoprotein fraction (primarily HDL) following the incubation of liposomal Nys than following the incubation of free Nys (Figure 5). There was a significant correlation between the lipoprotein lipid and protein profiles in human, dog and rat plasmas and the distribution of free Nys and liposomal Nys in plasma. Specifically, differences in the proportion of plasma lipoprotein cholesterol, triglyceride and apolar lipids (cholesteryl esters and triglycerides) carried by HDL influenced the distribution of free Nys and liposomal Nys within plasmas of different species. In a further study, we reported that as the amount of HDL protein decreased the amounts of free Nys and liposomal Nys recovered within this fraction decreased.103 Taken together these finding suggest that the distribution of Nys among plasma lipoproteins maybe defined by the proportion of lipid and protein carried by HDL, and may be an important consideration when evaluating the pharmacokinetics, toxicities and activities of these compounds following systemic administration to different animal species.
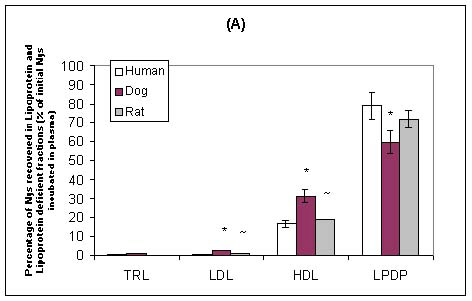
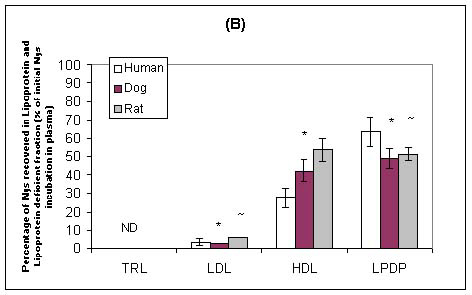
Figure 5: Distribution of free Nys (A) and liposomal Nys (B) (20mg/ml) in human, dog, and rat plasma following incubation for 5 minutes at 37oC. The data are expressed as percent of total Nys distributed in TRL, LDL, HDL, and LPDP fractions. The data are reported as means ± standard deviations (n = 3). * P < 0.05 compared to human plasma; ~ P < 0.05 compared to dog plasma.
Reference: 102. M. Ramaswamy, T. L. Wallace, P. A. Cossum and K. M. Wasan, Antimicrob. Agents Chemother. 43, 1424-8 (1999)
Clinical Studies
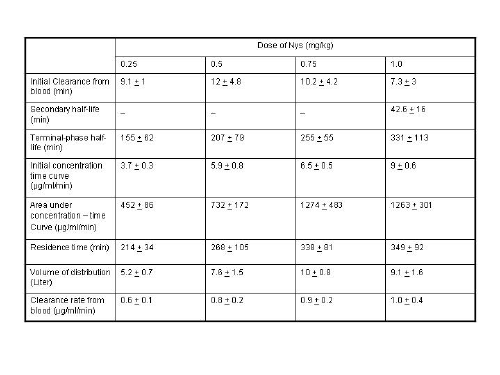
To date, few clinical studies with liposomal Nys have been reported. In 1993, Rios et al.104 reported on a phase I clinical trial that studied the safety, tolerance and pharmacokinetics of a single dose of liposomal Nys (Nyotran®) in seventeen patients with HIV infection. Four patient groups were studied, each at one of the following dose levels: 0.25, 0.5, 0.75, and 1 mg/kg. Patients were hospitalized during the first day of the study and then followed up as outpatients daily for 1 week. None of the patients have experienced any significant neurological, cardiac, pulmonary or renal toxic effects at any of the dose levels studied. Pharmacokinetic analysis (Table III) shows that after liposomal Nys is administered, the drug appears to distribute initially into the blood circulation and cleared with a terminal half-life (t1/2γ) of 5 hours. Furthermore, the kinetics of liposomal Nys appear to be dose independent in the range of 0.5 to 1 mg/kg body weight; however, the area under the concentration-time curve appears to increase in direct proportion to the dose administered for doses ranging from 0.25 mg/kg to 0.75 mg/kg, which suggests that in this dose range studied which suggests that in this dose range there is no apparent saturation of drug clearance mechanisms. However, area under the concentration-time curve does not increase when patients are administered 0.75 mg/kg versus 1.0 mg/kg suggesting a saturation of drug clearance once a dose of 0.75 mg/kg or above is administered. In addition, an increase in terminal half-life (t1/2γ) as dose increases further argues for saturation in drug clearance mechanisms. These results suggest that tissue exposure to liposomal Nys increases with increasing doses up to 0.75 mg/kg of this drug. With potentially enhanced tissue distribution, greater intracellular concentrations of Nys may be achieved.
Table 3: Pharmacokinetic parameters of liposomal Nys in patients with HIV infection.
Reference: 104. A. Rios, M. Rosenblum, G. Crofoot, R. P. Lenk, A. Hayman and G. Lopez-Berestein, J. Infect. Dis. 168, 253-4 (1993).
Recently, Krupova et al.105 reported the successful use of liposomal Nys in therapy of pulmonary aspergillosis refractory to conventional AmB therapy in cancer patients. However, more clinical studies with liposomal Nys are warranted.
Development Of Other Liposomal Polyene Antibiotics
A number of other liposomal polyene antibiotics have been developed. One of these compounds that have been incorporated into a lipid carrier is Hamycin. Hamycin has been used to treat a variety of yeast and other fungal infections through oral, topical and intraperitoneal routes. However, its parenteral use has been associated with high toxicity. In 1991, our group developed a multilamellar liposome formulation of Hamycin. The antifungal activity of Hamycin was maintained after liposome encapsulation and toxicity was reduced in vitro and in vivo as the concentration of cholesterol was increased.106 In additional, several studies by Moonis et al.107,108 reported the effectiveness of liposomal Hamycin in the treatment of experimental aspergillosis in Balb/c mice with limited side effects.
Recently, Harindran et al.109 reported the incorporation of a new polyene oxohexaene macrolide antibiotic isolated from Streptomyces CDRIL-312, HA-1-92, into liposomes containing phosphatidylcholine and cholesterol. The liposomal incorporated HA-1-92 resulted in considerably decreased toxicity when compared with free HA-1-92 in mice. Furthermore, liposomal HA-1-92 showed improved pharmacokinetic profiles in rats, in particular, a longer circulating half-life. When administered to aspergillosis- and cryptococcosis-infected Balb/c mice, liposomal HA-1-92 showed increased antifungal activity compared with free HA-1-92, improved survival rate and decreased colony-forming units in lung, liver, spleen, and kidney. Although these findings are promising additional studies are required.
SUMMARY
Fungal infections are on the rise worldwide, particularly as the population of immunocompromised patients continues to grow. By themselves, polyene antibiotics (i.e. nystatin and amphotericin B) are effective antifungal agents, though they are highly toxic. The goal of these liposomal formulations is to transport the drug through the body without exposing it to sensitive organs and tissues, and then to deliver it in concentrated doses to the target site. To a certain extent, these formulations accomplish this goal. For example, the maximum tolerable dose of AmB is about 1 mg/kg/day. However, lipid formulations of AmB allow physicians to go up to 5 times the dose of AmB without increasing infusion-related toxicities. These lipid formulations demonstrate improved efficacy, primarily because of the higher administered dose, and reduced kidney toxicity, compared to free form of the drug. As such, the future of liposomal polyene antibiotics is bright and it is apparent that these lipid-based products will replace the free form of these compounds as the mainstays in the treatment of systemic fungal infections.
ACKNOWLEDGMENTS
The authors would like to thank all the members of Drs. Wasan and Lopez-Berestein laboratories for their dedication and hard work in the development of ABLC and liposomal Nys. Much of the research from Drs. Wasan and Lopez-Berestein laboratories were funded from operating grants from the Canadian Institutes of Health Research, National Institutes of Health (US) and grant-in-aids from Aronex Pharmaceuticals Inc.
List Of Abbreviations
ABCD Amphotericin B Colloidal Dispersion
ABLC Amphotericin B Lipid Complex
AIDS Acquired Immunodeficiency Syndrome
AmB Amphotericin B
ATP Adenosine Triphosphate
DMPC Dimyristoyl Phosphatidylcholine
DMPG Dimyristoyl Phosphatidylglycerol
ND Not Detectable
Doc-AmB Amphotericin B formulated as a Micelle Suspension with Deoxycholate
DTNB Dithionitrobenzoate
HDL High Density Lipoproteins
HIV Human Immunodeficiency Virus
HPLC High Performance Liquid Chromatography
L-AmB Liposomal Amphotericin B
LDL Low Density Lipoproteins
LLC PK1 A pig kidney epithelial cell line
LPDP Lipoprotein Deficient Plasma
MIC Minimum Inhibitory Concentration
Nys Nystatin
RPMI 1640 A type of liquid cell culture medium
TRL Triglyceride Rich Lipoprotein
VLDL Very Low Density lipoproteins
REFERENCES
E. J. Anaissie, Opportunistic mycoses in the immunocompromised host: experience at cancer center and review, Clin. Infect. Dis. 14(suppl.1), 43-53 (1992).
M. A. Pfaller, R. Webzel, The impact of changing epidemiology of fungal infections in the 1990s. Eur. J. Clin. Microbiol. Infect. Dis. 11, 287-291 (1992).
M. D. Richardson, Opportunistic and pathogenic fungi, J Antimicrobial. Chemother. 28(suppl. A), 1-11 (1991).
T. J. Walsh, Invasive fungal infections: problems and challenges in developing new antifungal compounds in "Emerging Targets in Antibacterial and Antifungal" (J. Sutcliffe and N.H. Georgopapadakou, ed.), p. 249-373. Chapman & Hall, New York, 1992.
C. M. Beck-Sague, W. R. Jarvis, Secular trends in the epidemiology of nosocomial fungal infections in the United States, J. Infect. Dis. 167, 1247-51 (1993).
D. W. Denning, Epidemiology and pathogenesis of systemic fungal infections in the immunocompromised host, J. Antimicrob. Chemother. 28(suppl. B), 1-6 (1991).
R. D. Diamond, The growing problem of mycoses in patients infected with the human immunodeficiency virus, Rev. Infect. Dis. 13, 480-6 (1991).
G. P. Bodey. Fungal infection and fever of unknown origin in neutropenic patients, Am. J. Med. 80, 112-119 (1986).
M. D. Meyer, Current role of therapy with amphotericin B. Clin. Infect. Dis. 14, s154-160 (1992).
http://chem.sis.nlm.nih.gov/aidsdrg4.html
J. Bolard, How do the polyene macrolide antibiotics affect the cellular membrane properties?, Biochim. Biophys. Acta 864, 257-304 (1986).
N. H. Georgopapdakou and T. J. Walsh, Antifungal Agents: Chemotherapeutic Targets and Immunologic Strategies, Antimicrob. Agents Chemother. 40, 279-91 (1996).
J. Brajtburg, W. G. Powderly, G. S. Kobayashi and G. Medoff, Current understanding of mechanisms of action, Antimicrob. Agents Chemother. 34, 183-8 (1990).
D. W. Warnock, Amphotericin B: an introduction, J. Antimicrob. Chemother. 28, 27-38 (1991).
G. G. Chabot, R. Pazdur, F. A. Valeriote and L. H. Baker, Pharmocokinetics and toxicity of continuous infusion of amphotericin B in cancer patients. J. Pharm. Sci. 78, 307- 10 (1989).
J. P. Tolins and L. Raij, Adverse effect of amphotericin B administration on renal hemodynamics in the rat: neurohumoral mechanisms and influence of calcium channel blocker. J. Pharmacol. Exp. Ther. 245, 594-9 (1988).
M. L. Gardner, P. Godley and S.M. Wasan, Sodium loading treatment of amphotericin B-induced nephrotoxicity. DICP 24, 940-5 (1990).
R. R. New, M. L. Chance and S. Heath, Antileishmanial activity of amphotericin and other antifungal agents entrapped in liposomes, J. Antimicrob. Chemother. 8, 371-81 (1981).
J. R. Graybill, P.C. Craven, R. L. Taylor, D. M. Williams and W. E. Magee, Treatment of murine cryptococcosis with liposome-associated amphotericin B, J. Infect. Dis.145, 748-52 (1982).
G. Lopez-Berestein, R. Mehta, R. L. Hopfer, K. Mills, L. Kasi, K. Mehta, V. Fainstein, M. Luna, E. M. Hersh and R.L. Juliano, Treatment and prophylaxis of disseminated infection due to Candida albicans in mice with liposome-encapsulated amphotericin B, J. Infect. Dis. 147, 939-45 (1983).
G. Lopez-Berestein, R. L. Hopfer, R. Mehta, K. Mehta, E. M. Hersh and R. L. Juliano, Prophylaxis of Candida albicans infection in neutropenic mice with liposome-encapsulated amphotericin B. Antimicrob. Agents Chemother. 25, 366-77 (1984).
R. L. Hopfer, K. Mills, R. Mehta, G. Lopez-Berestein, V. Fainstein and R. L. Juliano, In vitro antifungal activities of amphotericin B and liposome-encapsulated amphotericin B. Antimicrob. Agents Chemother. 25, 387-9 (1984).
R. Mehta , G. Lopez-Berestein, R. Hopfer, K. Mills and R. L. Juliano, Liposomal amphotericin B is toxic to fungal cells but not to mammalian cells, Biochim Biophys. Acta 770, 230-4 (1984).
G. Lopez-Berestein, M. G. Rosenblum and R. Mehta, Altered tissue distribution of amphotericin B by liposomal encapsulation: comparison of normal mice to mice infected with Candida albicans, Cancer Drug Delivery 1, 199-205 (1984).
G. Lopez-Berestein, R. L. Hopfer, R. Mehta, K. Mehta, E. M. Hersh and R. L. Juliano, Liposome-encapsulated amphotericin B for treatment of disseminated candidiasis in neutropenic mice, J. Infect. Dis. 150, 278-83 (1984).
G. Lopez-Berestein, T. McQueen and K. Mehta, Protective effect of liposomal-amphotericin B against C. albicans infections in mice, Cancer Drug Delivery 2, 183-9 (1985).
G. Lopez-Berestein, V. Fainstein, R. Hopfer, K. Mehta, M. P. Sullivan, M. Keating, M.G. Rosenblum, R. Mehta, M. Luna, E. M. Hersh et al., Liposomal amphotericin B for the treatment of fungal infections in patients with cancer: a preliminary study, J. Infect. Dis. 151, 704-10 (1985).
V. J. Wiebe and M. W. De Gregorio, Liposome encapsulated amphotericin B: a promising new treatment for disseminated fungal infections, Rev. Infect. Dis. 10, 1097-101 (1988).
C. Tremblay, M. Barza, C. Fiore and F. Szoka, Efficacy of liposome-intercalated amphotericin B in the treatment of systemic candidasis in mice, Antimicrob. Agents Chemother. 26,170-3 (1984).
C. Tremblay, M. Barza, F. Szoka, M. Lahav and J. Baum, Reduced toxicity of liposome-associated amphotericin B injected intravitreally in rabbits, Invest. Opthal. Vis. Sci. 26, 711-8 (1985).
R. L. Taylor, D. M. Williams, P. C. Craven, J. R. Graybill, D. J. Drutz and W. E. Magee, Amphotericin B in liposomes: novel therapy of histoplasmosis, Am. Rev. Respir. Dis. 125, 610-16 (1982).
J. D. Berman, W. L. Hanson, W. L. Chapman, C. R. Alving and G. Lopez-Berestein, Antileishmanial activity of liposome-encapsulated amphotericin B in hamsters and monkeys, Antimicrobial. Agents Chemother. 30, 847-51 (1986).
C. B. Ponosian, M. Barza, F. Szoka and D. J. Wyler, Treatment of experimental cutaneous leishmaniasis with liposome-intercalated amphotericin B. Antimicrob. Agents Chemother. 25, 655-56 (1984).
S. L. Croft, R. N. Davidson and E. A. Thornton, Liposomal amphotericin B in the treatment of visceral leishmaniasis, J. Antimicrob. Chemother. 28, 111-8 (1991).
L. Gradoni, R. N. Davidson, S. Orsini and P. Betto, Activity of liposomal amphotericin B (AmBisome) against Leishmania infantum and tissue distribution in mice, J. Drug Targeting 1, 311-6 (1993).
G. Lopez-Berestein, G. P. Bodey, L. S. Frankel and K. Mehta, Treatment of hepatosplenic candidiasis with liposomal-amphotericin B, J. Clin. Oncol. 5, 310-7 (1987).
G. Lopez-Berestein, G. P. Bodey, V. Fainstein, M. Keating, L. S. Frankel, B. Zeluff, L. Gentry and K. Mehta, Treatment of systemic fungal infections with liposomal amphotericin B, Arch. Intern. Med.149, 2533-8 (1989).
J. Torre-Cisneros, J. L. Villanueva, J. M. Kindelan, R. Jurado and P. Sanchez-Guijo, Successful treatment of antimony-resistant visceral leishmaniasis with liposomal amphotericin B in patients infected with human immunodeficiency virus, Clin. Infect. Dis. 17, 625-7 (1993).
T. Fusai, R. Durand, Y. Boulard, M. Paul, C. Bories, D. Rivollet, R. Houin and M. Deniau, Importance of drug carriers in the treatment of visceral leishmaniasis, Medecine Tropicale 55, 73-8 (1995).
- Efficacy and safety of amphotericin B lipid complex injection (ABLC) in solid-organ transplant recipients with invasive fungal infections, Clin. Transplant. 14(4 Pt 1), 329-39 (2000).
P. Linden, P. Williams and K. M. Chan,
- A pharmacokinetic study of amphotericin B lipid complex injection (Abelcet) in patients with definite or probable systemic fungal infections, Antimicrob. Agents Chemother. 44(10), 2900-2 (2000).
A. Adedoyin, C. E. Swenson, L. E. Bolcsak, A. Hellmann, D. Radowska, G. Horwith, A. S. Janoff and R. A. Branch,
G. Lopez-Berestein, L. Kasi, M. G. Rosenblum, T. Haynie, M. Jahns and H. Glenn, Clinical pharmacology of 99mTc-labeled liposomes in patients with cancer, Cancer Res. 44, 375-8 (1984).
L. P. Kasi, G. Lopez-Berestein, K. Mehta, M. Rosenblum, H. J. Glenn, T. P. Haynie, G. Mavligit and S. M. Hersh, Distribution of pharmacology of intravenous 99mTc-labeled multilamellar liposomes in rats and mice, Int. J. Nucl. Med. Biol. 11, 35-7 (1984).
R. Perez-Soler, G. Lopez-Berestein, L. Kasi, F. Cabanillas, M. Jahns, H. Glenn, E. M. Hersh and T. Haynie, Distribution of technetium-99m-labeled multilamellar liposomes in patients with Hodgkins disease, J. Nucl. Med. 26, 743-7 (1985).
R. T. Mehta, T. J. McQueen, A. Keyhani and G. Lopez-Berestein. Phagocyte transport as mechanism for enhanced therapeutic activity of liposomal amphotericin B. Exp. Chemother. 40, 256-2 (1994).
K. M. Wasan, K. Vadiei, G. Lopez-Berestein and D. R. Luke. Pharmacokinetics, tissue distribution, and toxicity of free and liposomal amphotericin B in diabetic rats, J. Infect. Dis. 161, 562-6 (1990).
A. J. Atkinson and J. E. Bennett, Amphotericin B pharmacokinetics in humans, Antimicrobial. Agents Chemother. 13, 271-8 (1978).
et al., Liposomal amphotericin B (AmBisome) in Mediterranean visceral leishmaniasis: a multi-centre trial , Quart. J. Med. 87, 75-81 (1994). R. N. Davidson, L. Di Martino, L. Gradoni, R. Giacchino, R. Russo, G. B. Gaeta, R. Pempinello, S. Scott, F. Raimondi, A. Cascio
G. Lopez-Berestein, Liposomes as carriers of antifungal drugs. Annals of the New York Academy of Sciences 544, 590-7 (1988).
K. M. Wasan and G. Lopez-Berestein, Targeted liposomes in fungi: modifying the therapeutic index of amphotercin B by its incorporation into negatively charged liposomes, J. Liposome Res. 5, 883-903 (1995).
J. Brajtburg, S. Elberg, J. Bolard and G. Medoff, Interaction of plasma proteins and lipoproteins with amphotericin B, J. Infect. Dis.149, 986-992 (1984).
T. E. Andreoli, The anatomy of amphotericin B-cholesterol pores in lipid bilayer membranes, Kidney Int. 4, 337-345 (1973).
M. H. Koldin, G. S. Kobayashi, J. Brajtburg and G. Medoff, Effects of elevation of serum cholesterol and administration of amphotericin B complexed to lipoproteins on amphotericin B-induced toxicity to rabbits, Antimicrobial. Agents Chemother. 28, 144-5 (1985).
K. J. Christansen, E. M. Bernard, J. W. M. Gold and D. Armstrong, Distribution and activity of amphotericin B in humans, J. Infect. Dis. 152, 762-5 (1985).
K. M. Wasan, G. A. Brazeau, A. Keyhani, A. C. Hayman and G. Lopez-Berestein, Role of liposome composition and temperature on the distribution of amphotericin B in serum lipoproteins, Antimicrobial. Agents Chemother. 37, 246-250 (1993).
Preferential distribution of amphotericin B lipid complex into human HDL3 is a consequence of high density lipoprotein coat lipid content, J. Pharm. Sci. 88, 1149-55 (1999). A. L. Kennedy and K. M. Wasan,
J. Babiak and L. L. Rudel, Lipoproteins and atherosclerosis, Bailliere's Clin. Endocrinol. Metab. 1, 515-21 (1987).
R. J. Cushley, W. D. Treleaven, Y. I. Parmar, R. S. Chana and D. B. Fenske, Surface diffusion in human serum lipoproteins, Biochem. Biophys. Res. Commun. 146, 1139-45 (1987).
W. K. Surewicz, R. M. Epand, H. J. Pownall and S. W. Hui, Human apolipoprotein A-I forms thermally stable complexes with anionic but not with zwitterionic phospholipids, J. Biol. Chem. 261, 16191-7 (1986).
R. L. Juliano, C. W. M. Grant, K. R. Barber and M. A. Kalp, Mechanism of the selective toxicity of amphotericin B incorporated into liposomes, Mol. Pharmacol. 31, 1-11 (1987).
A. S. Janoff, L. T. Boni, M. C. Popescu, S. R Minchey, P. R. Cullis, T. D. Madden, T. Taraschi, S. M. Gruner, E. Shyamsunder, M. W. Tate et al., Unusual lipid structures selective reduce the toxicity of amphotericin B, Proc. Natl. Acad. Sci. U.S.A. 85, 6122-6 (1988).
W. R. Perkins, S. R. Minchey, L. T. Boni LT, C. E. Swenson, M. C. Popescu, R. F. Pasternack and A. S. Janoff, Amphotericin B-phospholipid interactions responsible for reduced mammalian cell toxicity, Biochim. Biophys. Acta. 1107, 271-2 (1992).
H. J. Krause and R. L. Juliano, Interactions liposome-incorporated amphotericin B with kidney epithelial cells, Mol. Pharmacol. 34, 286-97 (1988).
V. Joly, S. J. Line, C. Carbon and P. Yeni, Interactions of free and liposomal amphotericin B with renal proximal tubular cells in primary culture, J. Pharmacol. Exp. Ther. 255, 17-22 (1990).
J. Brajtburg, S. Elberg, G. S. Kobayashi and G. Medoff, Effects of serum lipoproteins on damage to erythrocytes and Candida albicans cells by polyene antibiotics, J. Infect. Dis. 153, 623-6 (1986).
J. Barwicz, R. Gareau, A. Audet, A. Morisset, J. Villiard and I. Gruda, Inhibtion of the interaction between lipoproteins and amphotericin B by some delivery systems, Biochem. Biophys. Res. Commun.181, 722-6 (1991).
K. M. Wasan, M. G. Rosenblum, L. Cheung and G. Lopez-Berestein, Influence of lipoproteins on renal cytotoxicity and antifungal activity of amphotericin B, Antimicrob. Agents Chemother. 38, 223-7 (1994).
K. M. Wasan and J. S. Conklin, Enhanced Amphotericin B Nephrotoxicity in Intensive Care Patients with elevated low-density lipoprotein cholesterol, Clin. Infect. Dis. 24, 78-80 (1997).
S. Jullien, A. J. Vertut-Croquin, J. Brajtburg and J. Bolard, Circular dichroism for the determination of amphotericin B binding to liposomes, Anal. Biochem. 1972, 197-202 (1998).
J. Bolard, J. Legrand, F. Heitz and B. Cybulska, One-sided action of amphotericin B on cholesterol-containing membranes is determined by its self association in the medium, Biochem. 30, 5707-15 (1991).
S. Jullien, J. Brajtburg and J. Bolard, Affinity of amphotericin B for phosphatidylcholine vesicles as a determinant of the in vitro cellular toxicity of liposomal preparations, Biochim. Biophys. Acta 1021, 39-45 (1990).
K. M. Wasan and J. S. Conklin, Evaluation of renal toxicity and antifungal activity of free and liposomal amphotericin B following a single intravenous dose to diabetic rats with systemic candidasis, Antimicrob. Agents Chemother. 40, 1806-10 (1996).
K. M. Wasan, V. B. Grossie Jr. and G. Lopez-Berestein, Effects of Intralipid infusion on rat serum lipoproteins, Lab Animals 28, 138-142 (1994).
K. M. Wasan, V. B. Grossie Jr. and G. Lopez-Berestein, Concentrations in serum and tissue distribution of free and liposomal amphotericin B in rats on continuous Intralipid infusion, Antimicrob. Agents Chemother. 38, 2224-6 (1994).
Pharmacokinetics, distribution in serum lipoproteins and tissues, and renal toxicities of amphotericin B and amphotericin B lipid complex in a hypercholesterolemic rabbit model: single-dose studies, Antimicrob. Agents Chemother. 42, 3146-52 (1998). K. M. Wasan, A. L. Kennedy, S. M. Cassidy, M. Ramaswamy, L. Holtorf, J. W. Chou and P. H. Pritchard,
Amphotericin B lipid complex or amphotericin B multiple-dose administration to rabbits with elevated plasma cholesterol levels: pharmacokinetics in plasma and blood, plasma lipoprotein levels, distribution in tissues, and renal toxicities, Antimicrob. Agents Chemother. 45, 1184-91 (2001) M. Ramaswamy, K. D. Peteherych, A. L. Kennedy and K. M. Wasan,
Safety of aerosolized amphotericin B lipid complex in lung transplant recipients, Transplantation 72, 545-8 (2001). S. M. Palmer, R. H. Drew, J. D. Whitehouse, V. F. Tapson, R. D. Davis, R. R. McConnell, S. S. Kanj and J. R. Perfect,
Eradication of severe neonatal systemic candidiasis with amphotericin B lipid complex, Ann. Pharmacother. 35, 1032-6 (2001). D. C. Knoppert, H. E. Salama and D. S. Lee,
Efficacy of amphotericin B lipid complex in the treatment of invasive fungal infections in immunosuppressed paediatric patients, Eur. J. Clin. Microbiol. Infect. Dis. 20, 77-82 (2001). R. Herbrecht, A. Avurignon, E. Andres, R. Guillemain, A. Suc, D. Eyer, C. Pailler, V. Letscher-Bru, G. Levenger and G. Schaison,
Treatment of postkeratitis fusarium endophthalmitis with amphotericin B lipid complex, Cornea 19, 853-6 (2000). D. Goldblum, B. E. Frueh, S. Zimmerli and M. Bohnke,
Amphotericin B lipid complex for the treatment of recurrent blastomycosis of the brain in a patient previously treated with itraconazole, South Med. J. 94, 548-9 (2001). P. P. Cook,
D. D. Lasic, Mixed micelles in drug delivery, Nature 355, 279-80 (1992).
C. Gates and R. J. Pinney, Amphotericin B and its delivery by liposomal and lipid formulations, J. Clin. Pharmacy Ther.18, 147-53 (1993).
S. W. Sanders, K. N. Buchi, M. S. Goddard, J. K. Lang and K. G. Tolman, Single-dose pharmacokinetics and tolerance of cholesterol sulphate complex of amphotericin B administered to healthy volunteers, Antimicrob. Agents Chemother. 35:1029-34 (1991).
R. Chopra, S. Blair, J. Strang, P. Cervi, K. G. Patterson and A. H. Goldstone, Liposomal Amphotericin B (AmBisome) in the treatment of fungal infections in neutropenic patients, J. Antimicrob. Agents 28, 93-104 (1991).
O. Ringden, F. Meunier, J. Tollemar, P. Ricci, S. Tura, E. Kuse, M. A. Viviani, N. C. Gorin, J. Klastersky and P. Fenaux et al., Efficacy of amphotericin B (AmBisome) in the treatment of invasive fungal infections in immunocompromised patients, J. Antimicrob Agents 28, 73-82 (1991).
F. Meunier, H. G. Prentice and O. Ringden, Liposomal amphotericin B (AmBisome): safety data from a phase II/III clinical trial, J. Antimicrob. Agents 28, 83-91 (1991).
Comparison of amphotericin B lipid complex (ABLC) vs. ambisome in the treatment of suspected or documented fungal infections in patients with leukemia, Leuk Lymphoma 40, 511-20 (2001). R. V. Fleming, H. M. Kantarjian, R. Husni, K. Rolston, J. Lim, I. Raad, S. Pierce, J. Cortes and E. Estey,
H. R. Pace and S. I. Schantz, Nystatin (Mycostatin) in the treatment of monilial and nonmonilial vaginitis, JAMA 162, 268-71 (1956).
J. E. Stark, Allergic pulmonary aspergillosis successfully treated with inhalations of nystatin, Dis. Chest. 51, 96-9 (1967).
V.D. Newcomer, E. T. Wright, T. H. Sternberg, J. H. Graham, R. H. Wier, R. O. Egebery, Evaluation of Nystatin in the treatment of coccidioimycosis in man, in "Therapy of Fungal Disease" (T. H. Sternberg and V. D. Newcomer, ed.), p. 260-7. Little Brown, Boston, 1955.
M. C. Broughton, M. Bard and N. D. Lees, Polyene resistance ergosterol producing strains of Candida albicans, Mycoses 34, 75-83 (1991).
Formulation, toxicity, and antifungal activity in vitro of liposome-encapsulated nystatin as therapeutic agent for systemic candidiasis, Antimicrob. Agents Chemother. 31, 1897-900 (1987). R. T. Mehta, R. L. Hopfer, L. A. Gunner, R. L. Juliano and G. Lopez-Berestein,
Toxicity and therapeutic effects in mice of liposome-encapsulated nystatin for systemic fungal infections, Antimicrob. Agents Chemother. 31, 1901-3 (1987). R. T. Mehta, R. L. Hopfer, T. McQueen, R. L. Juliano and G. Lopez-Berestein,
Activity of liposomal nystatin against disseminated Aspergillus fumigatus infection in neutropenic mice, Antimicrob. Agents Chemother. 41, 2238-43 (1997). T. L. Wallace, B. Paetznick, P. A. Cossum, G. Lopez-Berestain, J. H. Rex and E. Anaissie,
Liposomal nystatin against experimental pulmonary aspergillosis in persistently neutropenic rabbits: efficacy, safety and non-compartmental pharmacokinetics. J. Antimicrob. Chemother. 43, 95-103 (1999). A. H. Groll, C. E. Gonzalez, N. Giri, K. Kligys, W. Love, J. Peter, E. Feuerstein, J. Bacher, S. C. Piscitelli and T. J. Walsh,
Safety and efficacy of multilamellar liposomal nystatin against disseminated candidiasis in persistently neutropenic rabbits, Antimicrob. Agents Chemother. 43, 2453-7 (1999). A. H. Groll, V. Petraitis, R. Petraitiene, A. Field-Ridley, M. Calendario, J. Bacher, S. C. Pliscitelli and T. J. Walsh,
Compartmental pharmacokinetics and tissue distribution of multilamellar liposomal nystatin in rabbits, Antimicrob. Agents Chemother. 44, 950-7 (2000). A. H. Groll, D. Mickiene, K. Werner, R. Petraitiene, V. Petraitis, M. Calendario, A. Field-Ridley, J. Crisp, S. C. Piscitelli and T. J. Walsh,
, Comparison of in vitro activity of liposomal nystatin against Aspergillus species with those of nystatin, amphotericin B (AB) deoxycholate, AB colloidal dispersion, liposomal AB, AB lipid complex, and itraconazole, Antimicrob. Agents Chemother 43, 1264-6 (1999). K. L. Oakley, C. B. Moore and D. W. Denning
D. W. Denning and P. Warn, Dose range evaluation of liposomal nystatin and comparisons with amphotericin B and amphotericin B lipid complex in temporarily neutropenic mice infected with an isolate of Aspergillus fumigatus with reduced susceptibility to amphotericin B, Antimicrob. Agents Chemother. 43, 2592-9 (1999).
A. J. Carrillo-Munoz, G. Quindos, C. Tur, M. T. Ruesga, Y. Miranda, O. Delvalle, P. A. Cossum and Wallace, In-vitro antifungal activity of liposomal nystatin in comparison with nystatin, amphotericin B cholesteryl sulphate, liposomal amphotericin B, amphotericin B lipid complex, amphotericin B desoxycholate, fluconazole and itraconazole, J. Antimicrob. Chemother. 44, 397-491 (1999).
M. Ramaswamy, T. L. Wallace, P. A. Cossum and K. M. Wasan, Species differences in the proportion of plasma lipoprotein lipid carried by high-density lipoproteins influence the distribution of free and liposomal nystatin in human, dog, and rat plasma, Antimicrob. Agents Chemother. 43, 1424-8 (1999)
S. M. Cassidy, F. W. Strobel and K. M. Wasan, Plasma lipoprotein distribution of liposomal nystatin is influenced by protein content of high-density lipoproteins, Antimicrob. Agents Chemother. 42, 1878-88 (1998).
A. Rios, M. Rosenblum, G. Crofoot, R. P. Lenk, A. Hayman and G. Lopez-Berestein, Pharmacokinetics of liposomal nystatin in patients with human immunodeficiency virus infection, J. Infect. Dis. 168, 253-4 (1993).
Y. Krupova, M. Mistrik, E. Bojtarova, D. Sejnova, I. Ilavska, V. Krcmery Jr., Liposomal nystatin (L-NYS) in therapy of pulmonary aspergillosis refractory to conventional amphotericin B in cancer patients, Support Care Cancer 9, 209-210 (2001).
R. T. Mehta, T. J. McQueen, A. Keyhani and G. Lopez-Berestein, Liposomal hamycin: reduced toxicity and improved antifungal efficacy in vitro and in vivo, J. Infect. Dis. 164, 1003-6 (1991).
M. Moonis, I Ahmad and B. K. Bachhawat, Liposomal hamycin in the control of experimental aspergillosis in mice: relative toxicity, therapeutic efficacy and tissue distribution of free and liposomal hamycin, Indian J. Biochem. Biophys. 29, 339-45 (1992).
M. Moonis, I. Ahmad and B. K. Bachhawat, Mannosylated liposomes as carriers for hamycin in the treatment of experimental aspergillosis in Balb/C mice, J. Drug Targeting 1, 147-55 (1993).
J. Harindran, K. K. Chakraborty and S. R. Naik, Preparation, relative toxicity and therapeutic efficacy in mice and rats of liposomal HA-1-92, a new oxohexaene polyene macrolide antibiotic, J. Pharm. Pharmacol. 51, 771-6 (1999).
Corresponding Author: Kishor M. Wasan, Associate Professor & Chair, Division of Pharmaceutics and Biopharmaceutics, Faculty of Pharmaceutical Sciences, The University of British Columbia, 2146 East Mall Avenue, Vancouver, British Columbia, Canada, V6T 1Z3. kwasan@interchange.ubc.ca
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps