J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 6(1):13-19, 2003
Drug release characteristics of lipid based benzoporphyrin derivative.
Rubinah K Chowdhary, Isha Shariff and David Dolphin1
Department of Chemistry, University of British Columbia, Vancouver, British Columbia, CanadaReceived 26 August 2002, Revised 28 November 2002, Accepted 02 January 2003
PDF version
Abstract
PURPOSE. The purpose of this study was to examine the transfer of verteporfin (BPDMA) from its lipid based formulation to serum proteins. METHODS. As a result of BPDMA being confined to the lipid phase, it was found that fluorescence from the photosensitizer was highly concentration quenched. This phenomenon was used to demonstrate rapid transfer of lipid-based drug to various plasma components such as albumin and lipoproteins. Gel electrophoresis was used to show transfer of drug to lipoproteins. RESULTS. Loss of fluorescence quenching showed rapid transfer of the drug from its lipid based formulation to serum proteins. Gel electrophoresis showed that both the drug and phospholipid components were transferred to the lipoprotein fraction concurrently. The electrophoretic mobility of plasma lipoproteins was increased as a result of their interaction with lipid-based BPDMA. It was also shown that the lipid-based structures were readily destabilized in the presence of relatively low concentrations of plasma, and that liposomes of this lipid composition were highly unlikely to be found intact in the circulation following intravenous injection. CONCLUSIONS. Verteporfin is rapidly transferred from its lipid based formulation to serum proteins. This rapid transfer, particularly to lipoproteins, provides a mechanism for its rapid delivery to cells.
Introduction
Photodynamic Therapy (PDT) has been found useful for managing a number of clinical disorders involving tissue hyper-proliferation and those involving hyper-vascularization (1). It is an approved treatment for solid tumors and age related macular degeneration. The general mode of treatment involves injection of the photosensitizing drugs, followed by their photoactivation in situ . These drugs tend to localize, with some selectivity, in abnormal tissues and light activation produces short-lived cytotoxic oxidative species, which can induce localized tissue damage.
Benzoporphyrin derivative (BPDMA, verteporfin) is a hydrophobic chlorin-like photosensitizer, which has been shown to be a highly effective in vivo , with little of the skin sensitivity associated with other photosensitizing drugs (2). It has a hydrophobic nature which ensures rapid cellular uptake and localization at critical intracellular membranous organelles. However, it also has a tendency to undergo self aggregation in aqueous media (3), which can severely limit drug bioavailability to biological systems. Aggregation is also known to severely curtail the photosensitizing properties of tetrapyrrolic drugs, and this can adversely affect drug pharmacokinetics and biodistribution patterns. It is therefore important to introduce BPDMA into the bloodstream in its monomeric form. BPDMA was therefore formulated in lipids, which provide an injectable hydrophobic vehicle for intravenous drug delivery.
The lipid phase composition is dimyristoyl phosphatidyl choline, DMPC (C14 0 ) and an unsaturated egg phosphatidyl glycerol (EPG) mixture (C14-18 0-4 ), into which BPDMA is incorporated. The verteporfin formulation is cryopreserved and can be reconstituted by addition of water prior to injection.
When the BPDMA is restricted to the lipid phase at a high concentration, it is not surprising that its fluorescence would be susceptible to quenching by neighboring photosensitizer molecules. Fluorescence was, therefore, barely detectable even in highly dilute aqueous suspensions of lipid-based BPDMA. Flow cytometry studies showed that addition of drug-free (placebo) formulations, of the same lipid composition, to lipid-based BPDMA resulted in a immediate dramatic enhancement of the average fluorescence signal from the lipid-based population without an increase in particle size. This suggested concentration quenching of BPDMA fluorescence was due to restricted intermolecular spacing in the lipid phase of the formulation. It also showed that lipid-based BPDMA was able to undergo rapid transfer to drug free lipid-based formulations in aqueous suspension. Steady state studies of the distribution of lipid-based BPDMA between various plasma components (4) have shown verteporfin to associate predominantly with the lipoprotein fraction on mixing with plasma components. However, it was not known how rapidly drug transfer took place. Furthermore, it was also unclear, following drug delivery to lipoproteins on intravenous injection, if any liposomes were present in the circulation in intact form. The following studies were performed to shed some light on these matters.
MATERIALS AND METHODS
Lipid based verteporfin (BPDMA) and placebo formulations were provided by QLT Inc., Vancouver, British Columbia. High and low density human lipoproteins were purchased from Sigma
Fluorescence studies. Fluorescence studies were carried out using an Aminco-Bowman Series 2 luminescence spectrophotometer in pulsed mode with the sample compartment thermostatted to 23°C. Lipid based BPDMA suspensions (2 mL) for fluorescence studies were diluted in iso osmolar 5% w/v sterile packed dextrose solution to give an absorbance of approximately 0.1±0.01 at the excitation wavelength 434 nm. All solutions were equilibrated to room temperature prior to use. Immediately following each addition, fluorescence (λ ex 434 nm, λ em 690 nm) of the suspension was determined with time, and equilibration allowed to take place before addition of subsequent aliquots.
Interphospholipid BPDMA transfer. A concentrated solution of placebo phospholipids (a drug-free lipid-based formulation, having the same lipid composition and size distribution) was prepared which had a lipid content of 6.6 mg/mL. Aliquots of the placebo (100 μL) were added, with rapid mixing by inversion of cuvette, to 3 mL lipid-based BPDMA suspension diluted to give A 434 = 0.1. Fluorescence (λ ex 434 nm) was scanned between 650 and 730 nm.
Extraction of lipid-based BPDMA by bovine serum albumin. A stock solution of bovine serum albumin (BSA) was prepared to a concentration of 10 mg/mL, and added in aliquots (100 μL) to 2 mL of a lipid-based BPDMA suspension (A 434 = 0.1), and fluorescence (λ ex 434 nm, λ em 690 nm) measured with time
Demonstration of binding of lipid-based BPDMA to albumin. The ability of various chromophores, including tetrapyrroles, to quench protein emission (predominantly of tryptophan) by energy transfer has been frequently used to study their interaction with proteins (3,5). This phenomenon was used to confirm transfer of BPDMA from the lipid phase to albumin. Emission (λ ex 290 nm, λ em 340 nm to give final A 434 =0.1) was followed as lipid-based BPDMA was added to a 0.7 mg/mL solution of albumin.
Determination of the extent of extraction of lipid-based BPDMA by fetal bovine serum. A time based fluorescence trace was obtained using 2 mL lipid-based BPDMA suspension prepared as described above. One hundred μL of fetal bovine serum was added to the suspension and rapidly mixed. To obtain the 100% fluorescence reading for BPDMA, the lipid phases were disrupted by addition of 10 μL of 10% v/v Triton X-100.
Agarose Gel Electrophoresis. Lipid-based BPDMA gave a highly mobile band on electrophoresis. Keeping the BPDMA concentration constant at 0.37 mg/mL, titrations were carried out over a range of human plasma concentrations between 0-50% v/v. Following mixing, the samples were incubated for 5 min before being placed, in duplicate, into the electrophoresis wells.
Gel Electrophoresis. Agarose gel (0.75%) was prepared by dissolving 1.95 g of agarose in 250 mL 1X Tris Borate buffer, pH 8, with warming and stirring. This was poured into the gel mold and allowed to harden over 1h. Gel was laid in the electrophoresis tank, covered with 1X TBE, and run at constant voltage (60 V) for 1.5 h. Gel was scanned using Deskscan.
Sudan Black Staining. The gel was fixed for 10 min in an aqueous solution containing 10% glacial acetic acid and 60% ethanol, rinsed, and dried at 100oC for 1 hr. Sudan Black stain was prepared according to the instructions given in the Beckman Paragon electrophoresis system manual for lipoproteins (P/N 655910). Paragon Lipo stain (7% w/w) was diluted to 0.07% for use (3 mL into 165 mL ethanol and 135 mL de-ionized water). The gel was soaked in working stain for 1.5 h. Destaining of the gel was carried out in 45% ethanol in distilled water over 2 days with several changes of destaining solution.
Sub-micron particle sizing. Particle size was measured by dynamic light scattering using a NICOMP 370 sub-micron particle sizer. This included an instrument performance check out using standard beads of known size. Twenty μL of reconstituted lipid-based BPDMA (1.54 mg/mL) were diluted into 0.8 mL of either 9.5% lactose or plasma. The final concentration of lipid-based BPDMA was 37 μg/mL in each case. Following a 5 min incubation, particle size distribution was determined. A control experiment using plasma alone was also carried out.
Results
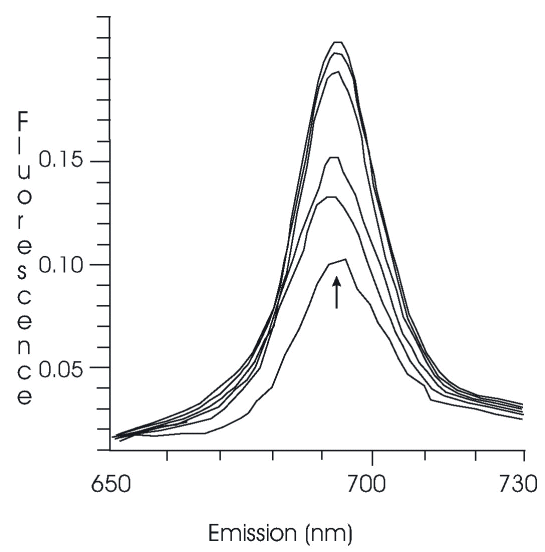
The lipid-based BPDMA suspension displays a constant, very low intensity emission. Fluorescence spectrophotometry scans (Figure 1) show dramatic increases in the 690 nm emission from aqueous lipid-based suspensions of BPDMA upon the addition of drug-free lipids. This demonstrates the ability of BPDMA to undergo rapid transfer between lipid phases. It also suggests one possible mechanism for drug transfer from the lipid-based formulation following injection into the blood stream.
Figure 1: Increase in fluorescence (λ ex 434 nm, λ em 690 nm) from a suspension of lipid-based BPDMA following addition of aliquots of drug-free lipid formulation.
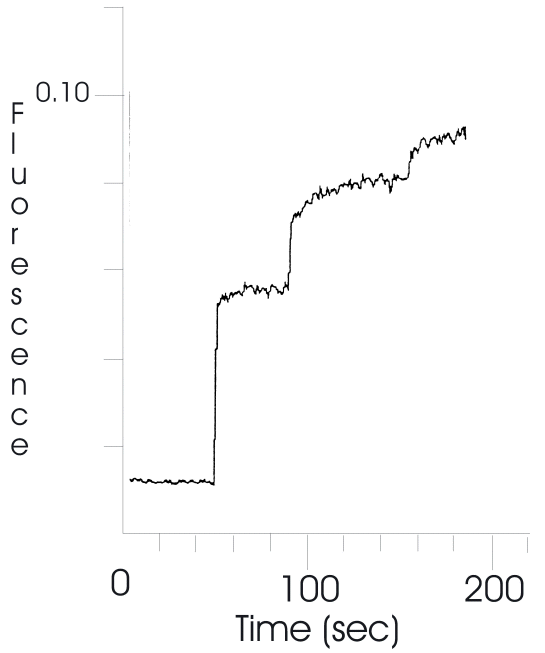
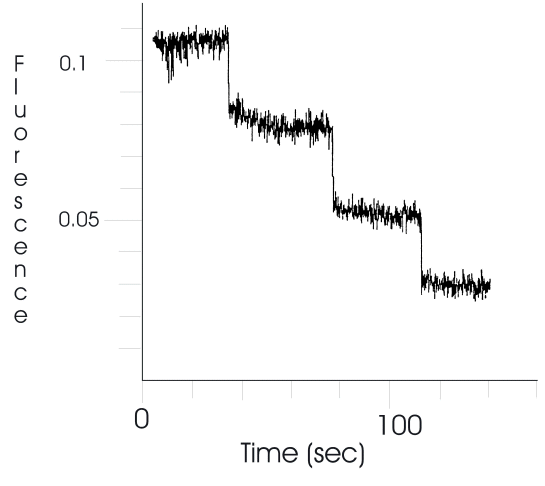
Similarly, albumin, the main plasma protein in circulation, was also found to be able to extract BPDMA from the lipid phase at concentrations considerably lower than those found under physiological conditions. Addition of albumin causes an increase in 690 nm fluorescence as a result of drug transfer out of the lipid phase (Figure 2), which results in the quenching of albumin emission at 294 nm due to binding of BPDMA to albumin (Figure 3). As both these events take place on a similar time scale, it can safely be deduced that the changes in emission reflect the rate and extent of drug transfer out of the lipid phase. A total of 140 μL (0.7 mg/mL albumin) was required for complete extraction of the BPDMA at A 434 =0.1.
Figure 2: Increase in fluorescence (λ ex 434 nm, λ em 690 nm) from a suspension of lipid-based BPDMA following addition of aliquots of bovine serum albumin.
Figure 3: Quenching of fluorescence (λ ex 290 nm, λ em 340 nm) of bovine serum albumin following binding of BPDMA.
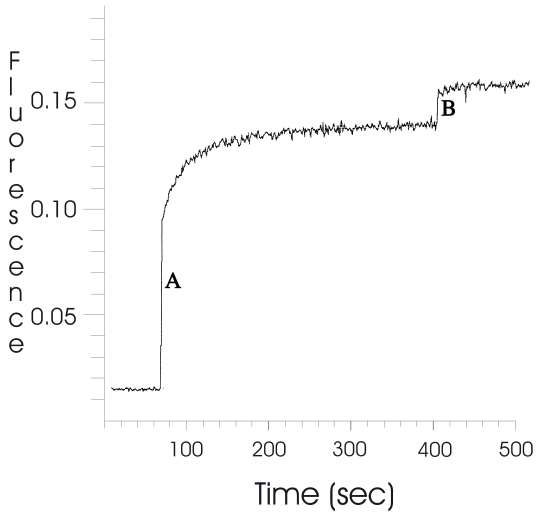
Addition of small amounts of purified human lipoproteins, LDL and HDL to suspensions of lipid-based BPDMA also resulted in instantaneous enhancement of emission at 690 nm. This suggests faster acquisition of drug compared to albumin, where 100% extraction took a few minutes. Similarly, addition of 5% v/v fetal bovine serum to a lipid-based BPDMA suspension was sufficient to attain almost complete extraction of BPDMA (Figure 4). Triton X-100 (0.5% v/v) was then used to disrupt the lipid phase after which the fluorescence signal in the presence of Triton X-100 was assumed to be equivalent to that of 100% drug release from the lipid phase. However, the fluorescence yield of BPDMA in the presence of 5%v/v FBS and Triton X-100 was found to be ~14% higher than in plasma alone. Conversely, in a solution containing BPDMA, phospholipids and Triton X-100, the fluorescence signal was reduced by the same amount on addition of 5% v/v FBS. Thus the difference in fluorescence signals was presumed to be due to different binding preferences of BPDMA in the two systems. Taking into account that the fluorescence of BPDMA in plasma is 14% higher in the presence of detergent, the 85% signal on addition of FBS to lipid-based BPDMA amounts to almost complete extraction of drug by the 5% v/v FBS. Thus extraction of lipid-based BPDMA can be achieved by considerably sub-physiological concentrations of albumin, lipoproteins or whole serum.
Figure 4: Extent of extraction of lipid-based BPDMA by 5% v/v fetal bovine serum by monitoring increase in fluorescence (λ ex 434 nm, λ em 690 nm). B. Addition of Triton X-100.
As lipoproteins are the main plasma components for transport of lipids and lipid soluble materials in the circulation, it is probable that interaction of lipid-based BPDMA with plasma lipoproteins results in the acquisition of phospholipids along with BPDMA. The extent of lipid acquisition could establish whether the lipid phases undergo destabilization as a consequence of their interaction with lipoproteins following injection. This, in turn, could determine whether these lipid phases circulate intact, when drug-depleted and whether they undergo size reduction due to partial loss of lipids, or whether they are absorbed in their entirety by the lipoprotein fraction.
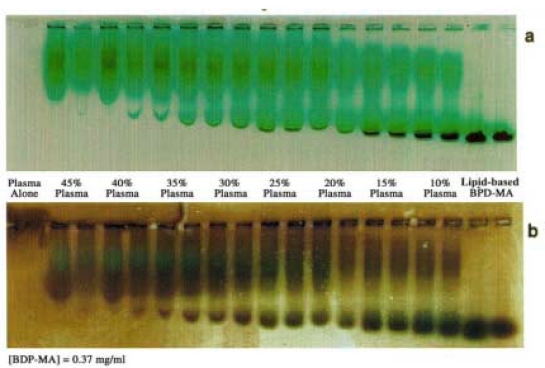
Some of these questions were addressed by the gel electrophoresis experiments. On account of the negatively charged head group of EPG and the carboxylate group of BPDMA, these lipid based particles give a highly mobile band on electrophoresis under physiological pH conditions, which was useful in examining their interaction with plasma components. The lipid-based BPDMA formulation itself consisted of at least 2 components with different electrophoretic mobilities; this could be due to differences in particle size and/or uneven distribution of charged components. The presence of non-liposomal material e.g. micellar structures formed predominantly from EPG/BPDMA is also likely, based on the negatively charged lipid head groups. Increasing the plasma concentration in a suspension of lipid-based BPDMA resulted in the gradual depletion and eventual disappearance of the lipid particles (Figure 5a). The decrease in this band is accompanied by a gradual decrease in the electrophoretic mobility, suggesting that zwitterionic DMPC might possibly lag marginally behind the drug and the negatively charged egg PG in terms of transfer to lipoproteins due to them both being negatively charged entities.
At the same time, the electrophoretic mobility of the lipoprotein band is also increased as the lipoprotein band acquires more of the lipid-based BPDMA. Figure 5b which highlights the lipid component by Sudan Black staining, confirms that the broad intermediate band corresponds to lipoproteins, and that lipids are also acquired by contact with increasing concentrations of lipoproteins. The lipid-based band is barely discernible in the presence of 35-45% human plasma. The BPDMA concentration in humans (0.37 mg/mL) is approximately 100-fold in excess of Cmax (maximum circulatory drug concentration) after intravenous injection of 0.3 mg/kg of BPDMA, with expected plasma concentration to be in the range of 4 μg/mL. Thus, it is highly unlikely that intact liposomes and other lipid-based structures could exist in the circulation following injection.
Figure 5: Agarose gel electrophoresis of lipid-based BPDMA showing assimilation of (a) liposomal BPDMA and (b) liposomal lipids (Sudan Black stained gel) into plasma lipoproteins.
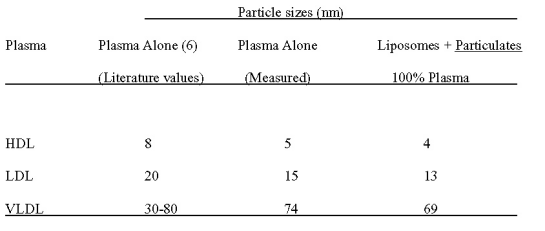
This observation was also confirmed by particle sizing experiments carried out in the presence of whole human plasma. Table 1 shows particle size analysis of BPDMA lipid-based suspensions before and after 5 min incubation in the presence of whole human plasma. Only particle sizes corresponding to lipoproteins were detectable when lipid-based BPDMA was exposed to human plasma. This confirms that liposomes do not remain intact in the circulation following injection. It is probable that any liposomal structure is rapidly destabilized on contact with plasma and the bilayer components as well as drug are incorporated into plasma lipoproteins.
Table 1: The effect of plasma on the size of BPDMA lipid-based formulations
DISCUSSION
The majority of work in the lipid-based/liposomal formulation area is aimed at providing controlled drug delivery. This is achieved either by triggering release of the therapeutic agent exploiting small differences in pH at target sites (7) e.g. lysosomes, inflamed tissue, or externally applied triggers e.g. light (8,9), heat (10), magnetic fields (11). Alternatively, the persistence time in the circulation is enhanced using components to minimize the interaction of plasma components with liposomes, such as reducing hydrophobicity of the liposomal surface (12). Such liposomes provide sustained release of the active agent over longer periods of time, and may also improve the safety of the treatment and selectivity at the target site.
In the case of lipid-based BPDMA, however, the aim is to rapidly deliver the drug to plasma lipoproteins since we have observed a correlation between efficient delivery of hydrophobic photosensitizers to lipoproteins and efficacy of PDT in vivo . Other researchers have also reported good results in vivo by pre-complexing hydrophobic drugs to lipoproteins, particularly LDLs (13-15). Performance depends on the interaction of proteins and lipoproteins in the circulation with the drug and/or the drug delivery vehicle. In the case of liposomes, lipid bilayer components play a critical part in determining the half-life of the drug in the circulation.
There are several characteristics of the lipid-based formulation discussed here that could explain the speed of drug delivery to plasma. The nature of the drug itself, the physico-chemical characteristics of the lipids, and the drug-lipid interaction within the lipid phases determines various characteristics such as the extent of drug loading achievable during formulation, behavior in the circulation e.g. lipid phase stability and drug release characteristics. Firstly, the unsaturated mixed egg phosphatidylglycerols contribute a high level of disorder to the lipid phases. It is known in liposomes that fluid bilayers are more easily destabilized in the presence of serum compared to those forming rigid, close packed bilayers e.g. liposomes prepared from saturated lipids or containing cholesterol (16). Secondly, the BPDMA lipid phases are composed of two classes of lipids (DMPC and EPG) with widely separated phase transition temperatures. This can create weak points at phase boundaries that are easily penetrated by proteins and therefore the system is more susceptible to disruption.
Fluorescence quenching studies have demonstrated rapid transfer of lipid-based drug to plasma proteins and lipoproteins, even at sub-physiological concentrations of plasma components. The gel electrophoresis experiments showed that as the lipid-based BPDMA is depleted of lipid by contact with increasing concentrations of plasma, it also displays a decrease in electrophoretic mobility. At the same time, the mobility of the lipoprotein band increases, presumably by acquisition of both BPDMA and lipids. Acquisition is complete at 35-45% v/v human plasma. Blood serum is generally considered highly liposomicidal, and this is largely attributed to the ability of lipoproteins to destabilize the liposomal bilayer.
As biological transporters of lipids in the circulation, it is perhaps inevitable that lipoproteins should readily and completely acquire phospholipids following injection. This process is greatly accelerated by the presence of phospholipid transferases in the circulation. The majority of interactions are likely to be with HDL, which can acquire up to 25% its own weight in lipids without displaying an increase in particle size, and reaching saturation at 0.25 mg phosphotidylcholine/mg (17). Blood concentration of these lipoproteins are 10 mg/mL, so it is not surprising that no intact liposomes or other lipid phases were detectable in the presence of plasma concentrations, even at excessively high lipid-based BPDMA concentrations.
In summary, the quenched fluorescence in lipid-based BPDMA enabled us to demonstrate the rapid speed of transfer of BPDMA from the lipid phase on contact with plasma. Gel electrophoresis experiments suggest that the transfer of lipids to lipoproteins takes place more or less concomitantly to that of BPDMA. They also demonstrate the complete disappearance of the lipid-based band with assimilation of both BPDMA and lipids into plasma lipoproteins. Sizing experiments also support the observation that intact liposomes and other lipid based structures will not be present in the circulation under physiological conditions. It is likely that the combination of high surface charge density and the dual lipid system of the lipid-based BPDMA formulation all contribute to the transient existence of the lipid-based BPDMA formulation in the circulation.
ACKNOWLEDGMENTS
We gratefully acknowledge financial support from NSERC in the form of a CRD grant, which allowed a mutually beneficial exchange of ideas between the Department of Chemistry, UBC and QLT Inc., Vancouver, B.C. The technical assistance of Valentina Zimina and Angela Cho of QLT Inc. is greatly appreciated.
REFERENCES
Pass H. I., Photodynamic therapy in oncology: mechanisms and clinical use. J. Natl. Cancer Inst. 85:443-456, 1993.
Richter, A.M., Jain, A.K., Canaan, A.J., Meadows, H. and Levy, J.G., Skin photosensitivity as a model in photodynamic therapy. Proceedings of SPIE p The Int. Soc. For Optical Engineering, 194-205, 1996.
Aveline B.M., Hasan, T. and Redmond, R.W., The effects of aggregation, protein binding and cellular incorporation on the photophysical properties of benzoporphyrin derivative monoacid ring A (BPDMA). J. Photochem. Photobiol. B, 30(2-3):161-169, 1995.
Allison B A., Pritchard, P.H., Richter, A.M. and Levy, J.G., The plasma distribution of benzoporphyrin derivative and the effects of plasma lipoproteins on its biodistribution. J. Photochem Photobiol., 52(3):501-507, 1990.
Epps D. E., Raub, T.J., Caiolfa, V., Chiari, A. and Zamai, M., Determination of the affinity of drugs toward serum albumin by measurement of the quenching of the intrinsic tryptophan fluorescence of the protein. J. Pharm. Pharmacol., 51(1):41-48, 1999.
Dobiasova M, Stribrna, J., Pritchard, P.H. and Frohlich, J.J., Cholesterol esterification rate in plasma depleted of very low and low density lipoproteins is controlled by the proportion of HDL2 and HDL3 subclasses: study in hypertensive and normal middle-aged and septuagenarian men. J. Lipid Res., 33(10):1411-1418, 1992.
Van Bambeke F, Kerkhofs, A., Schanck, A., Remacle, C., Sonveaux, E., Tulkens, P.M. and. Mingeot-Leclercq, M.P., Biophysical studies and intracellular destabilization of pH-sensitive liposomes. Lipids, 35(2):213-223, 2000.
Miller C.R., Clapp, P.J. and O'Brien, D.F., Visible light-induced destabilization of endocytosed liposomes. FEBS Lett., 467(1):52-56, 2000.
Chowdhary RK, Green, C.A. and Morgan, C.G., Dye-sensitized destabilization of liposomes bearing photooxidizable lipid head groups. J. Photochem. Photobiol., 58(3):362-366, 1993.
Ishida O, Maruyama, K., Yanagie, H., Eriguchi M. and Iwatsuru, M., Targeting chemotherapy to solid tumors with long-circulating thermosensitive liposomes and local hypothermia. Jpn. J. Cancer Res., 91(1):118-126, 2000.
Viroonchatapan E., Sato, H., Ueno, M., Adachi, I., Tazawa, K. and Horikoshi, I., Magnetic targeting of thermo sensitive magnetoliposomes to mouse livers in an in situ on-line perfusion system. Life Sci., 58(24):2251-2261, 1996.
Wollina U, T., Graefe and K. Karte, K., Treatment of relapsing or recalcitrant cutaneous T-cell lymphoma with pegylated liposomal doxorubicin. J. Am. Acad. Dermatol., 42:40-46, 2000.
Allison B. A., Waterfield, D., Richter, A.M., Levy, J.G., The effect of plasma lipoproteins on in vitro tumor cell killing and in vivo tumour photosensitization with benzoporphyrin derivative. J. Photochem. Photobiol., 54(5):709-715, 1991.
Hamblin, M.R. and Newman, E.L., On the machanism of the tumour-localising effect in photodynamic therapy. J. Photochem. Photobiol., 23(1):3-8, 1994.
Korbelik, M. Low density lipoprotein receptor pathway in the delivery of Photofrin: how much is it relevant for selective accumulation of the photosensitizer in tumors? J. Photochem. Photobiol., 12(1):107-109, 1992.
Allen T. M., A study of phospholipid interactions between high-density lipoproteins and small unilamellar vesicles. Biochim. Biophys. Acta, 640(2):385-397, 1981.
Lasic, D.D., Liposomes as a drug delivery system. In Liposomes: From Physics to Applications Chapter 11, Elsevier, New York, pp 265-321, 1993.
Corresponding Author: David Dolphin, Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia, Canada, V6T 1Z1. ddolphin@qlutinc.com
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps