J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 5(3):292-298, 2002
Hydroxypropyl-β-cyclodextrin-flutamide inclusion complex. II. Oral and intravenous pharmacokinetics of flutamide in the rat.
Zhong Zuo1, Yun K. Tam2, James Diakur and Leonard I. Wiebe3
Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, Canada1Present address: School of Pharmacy, Faculty of Medicine, The Chinese University of Hong Kong, Shatin, N.T., Hong Kong
2Kinetana Group Inc., 108 Advanced Technology Center, 9650 20th Avenue, Edmonton, Alberta, Canada
Received 24 October 2002, Revised 25 October 2002, Accepted 25 October 2002
PDF version
Abstract
PURPOSE: The objective of this work was to determine the pharmacokinetics of flutamide (FLT) and its active metabolite, 2-hydroxy-flutamide (FLT-2-OH) in rats, following formulation in hydroxypropyl-b-cyclodextrin (FLT-HPbCyD). METHODS: The pharmacokinetics of FLT-HPbCyD, FLT-suspension (FLT-SUSP), and FLT-solution (FLT-COSOLV) were compared after oral (p.o.) and intravenous (i.v.) administration, respectively. In a non-crossover design, male Sprague-Dawley rats received each formulation as a single oral dose [15 mg (54 mmol) FLT/kg] by oral gavage, or single i.v. dose [1.6 mg (5.8 mmol) FLT/kg] via an indwelling jugular vein catheter. FLT and its metabolite, FLT-2-OH, were determined in plasma and urine aliquots by an HPLC method. RESULTS: In a preliminary in vitro experiment, using the dialysis bag dissolution method, 80 % of a test dose of FLT was released from lyophilized FLT-HPbCyD into simulated gastric juice within 2 h, compared to less than 5% release from commercial FLT powder (FLT-SUSP). Following oral FLT-HPbCyD, the mean area under the plasma concentration curve (AUC0-¥) for FLT, was 1580 ± 228 ng.h/mL, with the maximum plasma concentration (Cmax; 1297 ± 127 ng/mL) at 0.5 h (Tmax) after administration. The AUC0-¥ and Cmax were significantly higher than after FLT-SUSP (AUC0-¥ 748 ± 206 ng.h/mL; Cmax 230 ± 111 ng/mL and Tmax 2.33 ± 0.29 h, respectively). After i.v. FLT-HPbCyD, the FLT AUC0-¥ was 1355 ± 162 ng.h/mL, compared to 1421 ± 283 ng.h/mL for FLT-COSOLV. FLT Cmax were 714 ± 144 mL/h and 735 ± 88 mL/h, respectively. The respective volumes of distribution (Vz) were 369 ± 191 mL and 242 ± 25 mL. The plasma concentration-time profile and pharmacokinetic parameters of FLT after FLT-HPbCyD and FLT-COSOLV did not differ significantly. The pharmacokinetic parameters for FLT-2-OH were formulation independent after i.v. dosing, but AUC0-¥; Cmax and Tmax, values were substantially greater with the FLT-HPbCyD in the oral study (40269 ± 5875 ng.h/mL, 4062 ± 502 ng/mL, and 3.50 ± 0.41 h, respectively). CONCLUSIONS: FLT from FLT-HPbCyD was released rapidly into solution in vitro and in vivo. FLT-HPbCyD improved oral bioavailability relative to FLT-SUSP. Intravenous pharmacokinetic profiles for both FLT and FLT-2-OH were identical following either FLT-HPbCyD or FLT-COSOLV, indicating that the FLT-HPbCyD formulation behaved as a true solution.
Introduction
Flutamide (4'-nitro-3'-trifluoromethylisobutyranilide; Euflex®, Drogenil®; FLT; MW 276) is a non-steroidal antiandrogen (1). It affects male secondary sex structures by inhibiting androgen uptake and/or inhibiting nuclear binding of androgens in target tissue, forming inactive complexes with nuclear androgen receptors, and reducing the rate of DNA synthesis in the prostate (2). The currently recommended dose is 250 mg orally, given three times daily. Higher doses do not appear to produce better therapeutic responses, and may be associated with a higher incidence and degree of gynecomastia (3), but low doses, which effect only partial blockage of the androgen receptor, can induce growth of tumor cells (4). FLT is commercially available as oral capsules and tablets, both with the same adult dose. A sustained-release dosage form has been investigated in an effort to enhance patients' compliance by lowering the number of tablets to be taken daily and to reduce the incidence of local side effects such as nausea and diarrhea (5).
In humans, FLT has low oral bioavailability due to extensive, rapid metabolic conversion (6). Six plasma metabolites have been identified, including 2-hydroxyflutamide (FLT-2-OH), the major metabolite (7). The clinical pharmacokinetics and pharmacodynamics of FLT have been reviewed (8, 9). The plasma half-lives of both FLT and FLT-2-OH in man are 5-6 h. Renal excretion of intact drug and metabolites accounts for approximately 28% of the dose within 24 h in rats (10), whereas male patients excrete 48% of the dose in 0-72 h urine, and % in 0-72 h feces (11). Long-term administration of the standard dose provides steady-state levels of FLT-2-OH over at least 18 months, indicating that changes in metabolic pattern are minimal during chronic use (12). In in vitro binding assays in androgen-sensitive tissues, FLT-2-OH is approximately 30 times more potent than FLT, but has only one percent of the potency of the natural agonist testosterone. Consequently, relatively high doses are required to ensure that FLT and/or FLT-2-OH plasma levels remain at least 500 to 1000-fold higher than circulating androgen in order to compete for receptor binding (13).
The low bioavailability of FLT from oral formulations may be due in part to poor wettability and low aqueous solubility, poor permeability, and rapid first pass hepatic extraction. We have postulated that solubility-related bioavailability deficiencies may be overcome by formulating FLT as a cyclodextrin complex. Cyclodextrins (CyDs) are cyclic α(1-4) linked oligosaccharides that have a hydrophilic external surface and a relatively hydrophobic interior surface, that can be exploited to solubilize hydrophobic drugs through the formation of inclusion complexes (14). In vivo dissociation of CyD complexes occurs rapidly, via simple diffusion-dissociation upon dilution, as well as through displacement by other hydrophobic compounds in the gastrointestinal tract (15).
The objectives of the current research were to formulate FLT in aqueous hydroxypropyl-β-cyclodextrin (HPbCyD), and to determine the pharmacokinetic parameters of FLT and its principle metabolite, FLT-2-OH, from FLT-HPbCyD. The data were compared to pharmacokinetic parameters for FLT and FLT-2-OH for suspension (FLT-SUSP) and solution (FLT-COSOLV) formulations.
MATERIALS AND METHODS
FLT, HPbCyD (average molar substitution 0.8), polyethylene glycol (Mn 200 Da) and dimethylsulfoxide (DMSO) were purchased from Sigma-Aldrich Canada Ltd. Other chemicals and solvents (analytical reagent grade), and distilled water (HPLC grade), were purchased from commercial suppliers. FLT-2-OH was a gift from Schering Plough Canada. Silastic® tubing (0.63 mm ID; 1.19 mm OD; Dow Corning Corp) and polyethylene tubing (PE50, I.D. 0.58 mm, O.D. 0.965 mm; Becton Dickinson) were used to catheterize the rats. Sterile Acrodisc® (0.2 mm) filtration was used to sterilize the intravenous (i.v.) formulations.
Dissolution of the two powder dosage forms (FLT and lyophilized FLT-HPbCyD; both <100 mesh) were studied in simulated gastric fluid. FLT powder (44.3 mg) or FLT-HPbCyD powder (containing the equivalent of 44.3 mg flutamide) were dispersed in dissolution medium (900 mL of simulated gastric fluid, pH 1.2; U.S.P.) previously equilibrated to 37 ± 0.5°C. The medium was stirred immediately at 50 r.p.m. Aliquots (1 mL) were withdrawn at 0.5, 1.5, 9, 24, 34, 64, 90, 120, 150 and 180 min, centrifuged and analyzed by HPLC on a Waters Radial PakÔ C18 10 mm reverse-phase column fitted with a mBondapakÔ C18 Guard-PakÔ guard column, using a mobile phase of methanol-water-glacial acetic acid-triethylamine (61/38/1/0.02 v/v/v/v) at 1.0 mL/min, with UV analysis at 300 nm (8). The cumulative dilution caused by sampling was corrected by immediately replacing 1 mL of the original medium after each sampling.
Male Sprague Dawley rats (350-400 g; Health Sciences Animal Service, University of Alberta) were used in accordance with guidelines of the Canadian Council on Animal Care. Experimental protocols were approved by the University of Alberta Health Sciences Animal Welfare Committee.
Three FLT dosage forms were used. The cyclodextrin inclusion complex formulation (FLT-HPbCyD) was prepared by adding excess FLT to a 50% w/v (50 mL) of HPbCyD in water. After vortex mixing (2 min), sonication (5 min), and shaking in water bath (25°C; 3 h), the solutions were filtered; the filtrates were lyophilized for storage at -20°C. This stock was reconstituted in water to a concentration of 5.3 mg/mL (19.2 mM) of flutamide for subsequent oral drug administration experiments and to a concentration of 2.0 mg/mL (7.1 mM) for i.v. drug administration. The FLT formulation (FLT-SUSP) was prepared with an aqueous vehicle containing carboxymethyl cellulose (0.5% w/v), polysorbate-80 (0.4% w/v), benzyl alcohol (0.9% v/v), and aqueous NaCl (0.9% w/v), to provide an FLT concentration of 24.5 mg/mL (88.7 mM). The FLT solution dosage form (FLT-COSOLV) was prepared by dissolving FLT in NaCl (0.9% w/v)-ethanol-PEG200 (2/1/3:v/v/v) to provide an FLT concentration of 24.5 mg/mL. The FLT-COSOLV and FLT-HPbCyD dosage formulations were sterile filtered through a 0.22 mm filter prior to injection.
For pharmacokinetic studies, rats (3 per group) were fasted overnight, then dosed with one of the three formulations, via either oral gavage (FLT-SUSP or FLT-HPbCyD; 15 mg/kg FLT) or i.v. injection (jugular vein catheter; FLT-HPbCyD or FLT-COSOLV; 1.60 mg/kg FLT). A jugular vein catheter, surgically inserted 48 h prior to dosing in order to minimize possible anesthetic effects, was used to withdraw blood for FLT and FLT-2-OH analysis by HPLC. Oral dose blood samples (0.1 mL) for FLT analysis were taken pre-dose, and at 0.17, 0.33, 0.5, 0.67, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5 and 4.0 h following injection, and additionally at 6, 8, 10, 15, 20 and 25 h for FLT-2-OH analysis; blood volume losses were compensated by injecting saline (0.1 mL) containing heparin (100 units/mL) via the sampling catheter. I.V. dosing blood specimens (0.2 mL) were withdrawn through the dosing catheter at 0.08, 0.17, 0.33, 0.5, 0.67, 1, 1.5, 2, 2.5, 3 and 3.5 h. After i.v. dosing via the catheter, the dose was followed by sequential injections of saline (0.1 mL), rat blood (0.1 mL) and saline (0.1 mL), each injection containing 100 unit/mL heparin. The i.v. protocol removed approximately twice the volume of blood over the `acute sampling phase' (pre-dose to 4 h) as did the oral regimen, but over the entire study, losses are similar because more samples were taken from the oral group. Although the net volume of blood removed from each of the oral and i.v. groups are approximately equal over 25 h, re-injection of blood (i.v. group) partially compensated for the early-times differences, thereby ensuring that physiological artifacts introduced by sampling would be relatively constant throughout this study. However, the main reason for injecting blood back through the catheter was to remove residual drug from the catheter after i.v. dosing through this catheter.
The whole blood samples (0.1 or 0.2 mL) were centrifuged to obtain plasma, which was analyzed for FLT and FLT-2-OH. The plasma was spiked with 2-hydroxy-5-nitro-benzaldehyde (internal standard; 25 mL; 1 mg/mL in methanol), then deproteinized by diluting with methanol (1 mL), vortex mixed (30 sec), chilled (4°C for 1 h) and centrifuged (800 x g) for 20 min). The supernatant was withdrawn and evaporated at room temperature under a stream of dry N2 at 25°C. The residue was dissolved in H2O-ethyl ether (1-10, v/v; 1 mL) and the organic layer was separated and evaporated. This final residue was dissolved in methanol (0.1 mL) for HPLC analysis (8). FLT recovery in the extraction step (90.9 ± 0.5% at 1 mg/mL) was determined by comparing the internal standard peak area of spiked samples with those obtained for an equivalent amount (in methanol) directly injected into the HPLC. The retention times of the internal standard, FLT-2-OH and FLT were 5.1, 6.2 and 8.6 min, respectively. Inter- and intra-assay variabilities were both less than 10%. The detection limit for FLT in methanol was determined to be approximately 5 ng/mL.
The plasma concentration data for FLT and FLT-2-OH were analyzed by computer. A non-compartmental model was applied. Each AUC0-¥ was obtained by a log-linear trapezoid method and the t1/2 (lambda Z) was determined based on the three terminal concentration-time points (WinNonlin® v. 1.1; Pharsight Corporation, USA). Significance of differences between experiments was calculated by Student's t test (unpaired) in SigmaPlot® v.4; RockWare Inc., USA), using two tailed distributions and a two-sample unequal variance (heteroscedastic) method. In all cases, statistical significance was determined at the 95% confidence level (p < 0.05).
Results
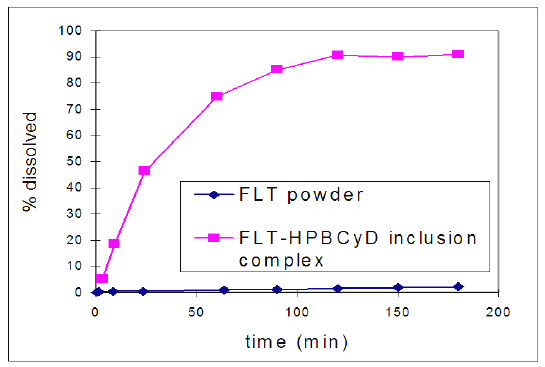
Dissolution and release of FLT from the lyophilized FLT-HPbCyD formulation was found to be much more rapid than dissolution of powdered FLT (Fig. 1) in artificial gastric juice.
Figure 1: Dissolution profiles for powdered FLT and powdered and FLT-HPbCyD (both <100 mesh) in simulated gastric fluid (pH 1.2; 37°C).
The observed increase in dissolution rate was attributed to improved wetting followed by increased dissolution of FLT from the FLT-HPbCyD complex, and to rapid equilibration of the FLT-HPbCyD formulation in the dissolution medium. HPLC analysis and mass balance studies of the medium during incubation showed no evidence of FLT degradation in simulated gastric juice, and there are no literature reports of non-metabolic degradation.
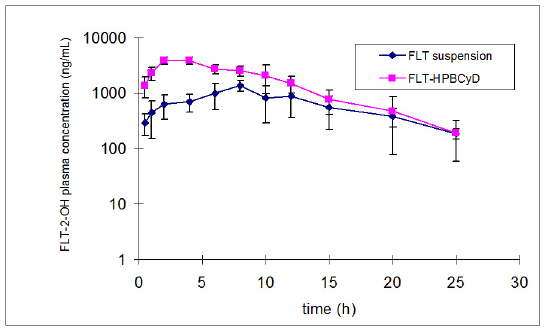
The mean plasma concentration data for FLT and FLT-2-OH over time after oral administration of FLT-SUSP and FLT-HPbCyD are presented in Figures 2 and 3.
Figure 2: Plasma concentration of FLT-2-OH after oral administration of the FLT-SUSP and FLT-HPbCyD dosage forms (FLT-equivalent dose = 15 mg/kg). Error bars delineate 1 S.D. from the mean, n = 3.
Figure 3: Pharmacokinetic data for FLT and FLT-2-OH after oral administration of FLT-SUSP and FLT-HPbCyD dosage forms (FLT-equivalent dose = 15 mg/kg; n = 3 per group).
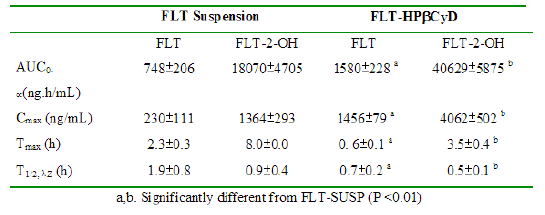
The mean area under the plasma concentration curve for FLT (AUC0-¥ 1580 ± 228 ng.h/mL), and the FLT Cmax (1456 ± 79 ng/mL) after the FLT-HPbCyD dose were much higher than after the FLT-SUSP dose (748 ± 206 ng.h/mL and 230 ± 111 ng/mL, respectively). Moreover, Cmax appeared at shorter times (Tmax) after dosing with FLT-HPβCyD (0.56 ± 0.10 h) than after FLT-SUSP (2.3 ± 0.3 h; P ≤ 0.0005). The relative bioavailability [F = (AUC0-¥(HPbCyD)/AUC0-¥(FLT-SUSP))*100; equal doses] for FLT from the FLT-HPbCyD formulation was 211%, and 224% for the metabolite. The calculated pharmacokinetic parameters are summarized in Table 1.
Table 1: Pharmacokinetic data for FLT and FLT-2-OH after oral administration of FLT-SUSP and FLT-HPbCyD dosage forms (FLT-equivalent dose = 15 mg/kg; n = 3 per group).
In the i.v. experimental protocol, the jugular vein catheter was used as both the dosing site and the sampling site because of the relative ease of maintaining the patency of a single indwelling catheter. Minimal catheter-derived cross-contamination from dose to first sample was confirmed experimentally. Ex vivo catheter washing experiments showed that after injection of either dosage formulation, a single saline wash would leave approximately 0.1% of the injected dose (640 mg) in the catheter, which, if totally recovered in the first blood sample (0.2 mL), would produce an apparent concentration of approximately 3200 ng/mL. In a preliminary animal experiment in which the dose was followed by a single saline wash (0.1 mL), the first blood contained around 9000 ng/mL, more than 30% of which could possibly be attributed to cross-contamination from the administered dose. When a whole blood rinse and a second saline push followed the initial saline wash in vitro, no FLT was detectable in the final saline rinse. The single-catheter protocol was therefore deemed appropriate for the i.v. pharmacokinetic experiments.
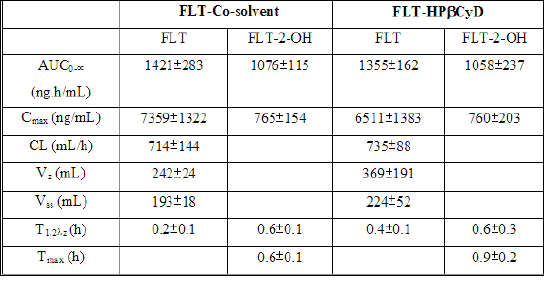
The initial FLT concentrations determined 0.08 h after i.v. dose were 3781 ± 494 ng/mL after FLT-HPbCyD (Fig. 4) and 3907 ± 388 ng/mL after the FLT-COSOLV dose (Fig. 5). FLT concentrations decreased rapidly, with respective half-lives (T1/2λz) of 0.4 ± 0.1 h and 0.2 ± 0.1 h. These values do not differ statistically (P = 0.37). The pharmacokinetic parameters (Table 2) for i.v. doses of FLT-HPbCyD were also not significantly different for the FLT-COSOLV dose. The calculated AUC0-∝ for FLT after FLT-HPbCyD and FLT-COSOLV, respectively, were 1355 ± 162 ng.h/mL and 1421 ± 283 ng.h/mL. The respective volumes of distribution (Vz) for FLT were 369 ± 191 mL and 242 ± 24 mL, and the clearances were 735 ± 88 mL/h and 714 ± 144 mL/h, respectively.
Table 2: Pharmacokinetic parameters for FLT and FLT-2-OH in rats, after single i.v. doses of either FLT-COSOLV or FLT-HPbCyD (FLT-equivalent dose = 1.6 mg/kg; n = 3 per group)
Figure 4: Plasma concentrations of FLT (ng/mL) following i.v. administration of the FLT-HPbCyD and FLT-COSOLV dosage forms (FLT-equivalent dose = 1.6 mg/kg). Error bars delineate 1 S.D. from the mean, n = 3.
The levels of FLT-2-OH reached peak values of 760 ± 203 ng/mL at 0.6 ± 0.1 h after i.v. injection of FLT-HPbCyD, and 765 ± 154 ng/mL at 0.9 ± 0.2 h after injection of the FLT-COSOLV formulation. The FLT-2-OH AUC0-¥ after FLT-HPbCyD was 1058 ± 237 ng.h/mL, not significantly different (P = 0.9) from that following the FLT-COSOLV dose (1076 ± 115 ng.h/mL). The absolute bioavailability for FLT with the FLT-HPbCyD formulation is 0.12, calculated from AUC and dose data in Tables 1 and 2.
Figure 5. Plasma concentrations of FLT-2-OH (ng/mL) following an i.v. dose of the FLT-HPβCyD and FLT-COSOLV dosage forms (FLT-equivalent dose = 1.6 mg/kg). Error bars delineate 1 S.D. from the mean, n = 3
Discussion
Native CyDs, and especially their more water-soluble derivatives (e.g. HPbCyD), have traditionally been used to increase oral bioavailability by increasing the dissolution of a given drug (15). Such applications have been successful when the rate-limiting step in drug absorption is dissolution of the drug itself and not absorption across the intestinal mucosa (14). It has recently been shown that FLT is rapidly absorbed by passive transcellular diffusion, a process that actually decreases at higher concentrations of HPbCyD. The latter phenomenon was attributed to lower availability of FLT for absorption because of a greater degree of complexation as the concentration of HPbCyD increased (16). These findings, together with current dissolution and pharmacokinetic data, support an FLT-HPbCyD absorption model based on increased FLT solubility. Increased FLT dissolution in aqueous HPbCyD would lead to efficient passive absorption by producing high concentrations of free FLT adjacent to the mucosal lining. In this paradigm, FLT-HPbCyD produces higher free FLT concentrations than concentrations derived from the FLT-SUSP formulation. The oral dose pharmacokinetic parameters for both FLT and FLT-2-OH show that the FLT-HPbCyD formulation provides more extensive and more rapid absorption. Moreover, the higher plasma levels and greater AUC of FLT from FLT-HPbCyD translate into proportional increases in the AUC of the active metabolite FLT-2-OH. This implies that this metabolic route is not saturated under these conditions, and indicates that low bioavailability from the oral FLT-SUSP is not attributable solely to a first pass metabolism phenomenon. The FLT-SUSP and FLT-HPbCyD dosage forms were administered in different liquid bases which arguably could affect their absorption; FLT-SUSP in a complex mixture that could possibly have acted as a partial solvent, and FLT-HPbCyD in aqueous solution. Co-solvents (co-solvent effects) usually improve absorption if drug wetting and dissolution are poor, but in this case, there is no evidence of such effect. Of course, the possibility remains that the liquid vehicle impaired FTL absorption from the FLT-SUSP dose, but intuitively this would not be highly probable. Since the formulations have varying properties, dosing artifacts (e.g. deposition of sticky or particulate formulations in the esophagus) could conceivably arise. However, no evidence of dose-loss during dosing was observed, and data support uniform dosing among members of each group, if not uniformity among the groups.
The pharmacokinetics of FLT and FLT-2-OH after the FLT-solution doses (FLT-HPbCyD and FLT-COSOLV) is virtually identical, implying rapid release of FLT from FLT-HPbCyD through dilution. It has been reported that only the earliest pharmacokinetic time points will be perturbed by CyD complexation, and then only for strongly bound (K > 105 M-1) drugs (17, 18). The stability constant of FLT-HPbCyD (356 M-1 at room temperature) (16) indicates relatively weak binding that will lead to rapid and complete release of the FLT on dilution. Intuitively, one might anticipate that precipitation would occur upon dissociation of a poorly water-soluble drug (e.g. FLT) from a CyD complex, but changes in the free: complexed ratio of a sparingly water-soluble drug have been shown to depend on the phase-solubility behavior of the system. Therefore, if the CyD complex represents a 1:1 ratio of drug and host, there will be a linear increase in drug solubility as the CyD concentration increases. Dilution of the drug-CyD complex releases the drug, but the drug will not precipitate regardless of the extent of dilution even if the concentration of drug exceeds its water-solubility limit (17). In other words, precipitation of FLT could occur on dilution if there were a nonlinear relationship between drug solubility and cyclodextrin concentration. However, in the case of FLT-HPbCyD, a 1:1 complex with linear solubility properties is formed (16), so no precipitation would be anticipated. On the other hand, co-solvent formulations based on alcohols and glycols will increase the solubility of a poorly water-soluble drug in a non-linear fashion with respect to co-solvent concentration. Precipitation may occur in such systems at any dilution where the equilibrium drug solubility is lower than the dilution concentration line at a given CyD concentration (15). The experimental data for the FLT-COSOLV formulation used in this study did not show any indication of precipitation upon i.v. injection (e.g., the Cmax was identical for both dosage forms). The FLT-COSOLV formulation is unsuitable because precipitation could still occur during storage, and the high percentage of alcohol in the co-solvent is inappropriate for a clinical dosage form.
In summary, the FLT-HPbCyD formulation of FLT has been shown to double the relatively low oral bioavailability of FLT in rats. The in vitro solubility and dissolution rate of FLT from FLT-HPbCyD, together with increased relative oral bioavailability of FLT from FLT-HPbCyD inclusion, provided the rationale for i.v. dosing studies with FLT-HPbCyD. Intravenous pharmacokinetic parameters for FLT and its biologically active metabolite (FLT-2-OH), following FLT-HPbCyD dosing, were identical to the pharmacokinetic parameters for the FLT-COSOLV dosage form. The validity of the rat as a model for human FLT absorption has not been fully established. However, the Tmax for absorption of FLT from FLT-SUSP (2.3 h) compares favorably with human data (~2 h) reported in the literature (8). That study also reported a 6 h Tmax for FLT-2-OH, compared to 3-5 h (Fig. 3) now reported for the rat model. These data could rationalize the development of a parenteral delivery form for FLT. More realistically, however, would be the development of an oral FLT-HPbCyD dosage form to increase both the oral bioavailability of FLT and the AUC of the biologically active metabolite FLT-2-OH. Additionally, the lyophilized FLT- HPbCyD powder may be suitable for long tern storage, and can readily be reconstituted to the desired concentration just prior to use.
Acknowledgements
Z. (Joan) Zuo was the recipient of pre-doctoral fellowships from PMAC/MRC of Canada, and The University of Alberta. This research was supported in part through the Medical Research Council of Canada (grant 13480).
a, a,a-trifluoro-2-methyl-4-nitro-m-propionotoluidide (flutamide), in men following a single oral 200 mg dose. J. Clin. Endocrinol. Metab. 41: 373-379.
Baker, J. W., Bachman, G. L., Schumacher, I., Roman, D. P., Tharp, A. (1967). Synthesis and bacteriostatic activity of some nitrotrifluorometylanilides. J. Med. Chem. 10: 93-95.
Neri, R. (1989). Pharmacology and pharmacokinetics of flutamide. Urology (Supplement) 34: 19-21.
Airhart, R. A., Barnett, T. F., Sullivan, J. W., Levine, R. L., Schlegel, J. U. (1978). Flutamide therapy for carcinoma of the prostate. South. Med. J. 71, 798-803.
Labrie F., Dupont, A., Belanger, A. (1985). Complete androgen blockade for the treatment of prostate cancer. In: De Vita, V. T., Hellman, S., Rosenberg, S. A. (eds) Important Advances in Oncology, J. B. Lippincott, Philadelphia, pp 193-217.
Asade, R. H., Prizont, L., Muino, J. P., Tessler, J. (1991). Steady-state hydroxyflutamide plasma levels after the administration of two dosage forms of flutamide. Cancer Chemother. Pharmacol. 27: 401-405.
Radwanski, E., Perentesis, G., Symchowicz, S., and Zampaglione, N. (1989). Single and multiple dose pharmacokinetic evaluation of flutamide in normal geriatric volunteers. J. Clin. Pharmacol. 29: 554-558.
Berson, A., Wolf, C., Chachaty, C., Fisch, C., Fau, D., Eugene, D., Loeper, J., Gauthier, J-C., Beaune, P., Pompon, D., Maurel, P., Pessayre, D. (1993). Metabolic activation of the nitroaromatic antiandrogen flutamide by rat and human cytochromes P-450, including forms belonging to the 3A and 1A subfamilies. J. Pharmcol. Exp. Ther. 265: 366-372.
Schulz, M., Schmoldt A., Donn, F., Becker H. (1988). The pharmacokinetics of flutamide and its major metabolites after a single oral dose and during chronic treatment. Eur. J. Clinical Pharmacol. 34:633-636.
Brogden, R. N., Clissold, S. P. (1989). Flutamide: a preliminary review of its pharmacodynamic and pharmacokinetics properties, and therapeutic efficacy in advanced prostatic cancer. Drugs 38: 185-203.
Neri, R., Florance, K., Koziol, P, Van Cleave S. (1972). A biological profile of a non-steroidal antiandrogen, SCH 13521 (4-nitro-3-trifluoromethyl-isobutyranalide). Endocrinology 91, 427-437.
Katchen, B., and Buxbaum, S. (1975). Disposition of a new, nonsteroid, antiandrogen,
Sogani, P. C., Vagaiwala, M. R., Whitmore, W. F. (1984). Experience with flutamide in patients with advanced prostatic cancer without prior endocrine therapy. Cancer 54: 744-750.
13. Simard, J., Luthy, I., Guay, J., Belanger, A., Labrie, F. (1986). Characteristics of interaction of the antiandrogen flutamide with the androgen receptor in various target tissues. Mol. Cell. Endocrinol. 44: 261-270.
Szejtli, J. (1994). Medicinal applications of cyclodextrins. Med. Res. Rev. 14, 353-386.
bCyD with bile salts in aqueous solutions. Chem. Pharm. Bull. 34: 1395-1398. Miyajima, K., Yoko, M., Komatsu, H., Nakagaki, M. (1986). Interaction of
Rajewski, R. A., Stella, V. J. (1996). Pharmaceutical application of cyclodextrins. 2. In vivo drug delivery. J. Pharm. Sci. 85, 1142-1169.
b-cyclodextrin complex: formation, physical characterization and absorption studies using the Caco-2 in vitro model. J. Pharmacy Pharm. Sci. 3: 220-227. Zuo, Z., Kwon, G., Diakur, J., Stevenson, B., Wiebe, L. I. (2000). Flutamide hydroxypropyl-
Stella, V. J., Rajewski, R. A. (1997). Cyclodextrins: their future in drug formulation and delivery. Pharm. Res. 14: 556-567.
Irie, T., Uekama, K. (1997). Pharmaceutical applications of cyclodextrins. III. Toxicological issues and safety evaluation. J. Pharm. Sci. 86:147-162.
Corresponding Author: Leonard I. Wiebe, 3118 Dentistry/Pharmacy Centre, University of Alberta, Edmonton, Alberta, Canada, T6G 2N8. leonard.wiebe@ualberta.ca
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps