J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 5(3):205-212, 2002
Reporting degree of deacetylation values of chitosan: the influence of analytical methods.
Tanveer Ahmad Khan
Kulliyyah of Pharmacy, International Islamic University Malaysia, 25710 Kuantan. Malaysia.Kok Khiang Peh1
School of Pharmaceutical Sciences, University of Science Malaysia, 11800 Penang, MalaysiaHung Seng Ch'ng
Hunza Labs Ltd., Innovation and Consultancy Centre, University of Science Malaysia, 11800 Penang, Malaysia.Received 22 August 2001, Revised 9 February 2002, Accepted 21 August 2002
PDF version
Abstract
PURPOSE: To investigate and compare the effect of three analytical methods, hydrogen bromide titrimetry (HBr titrimetry), infrared spectroscopy (IR spectroscopy), and first derivative UV-spectrophotometry (FDUV-spectrophotometry) in the determination of degree of deacetylation (DD) of chitosan. METHODS: Three different chitosan samples were selected for the DD quantification employing HBR titrimetry, IR spectroscopy with samples in the forms of KBr disc (at ratios of 1:2 and 1:3) and thin film (concentrations of 0.5 percent and 1 percent), and FDUV- spectrophotometry. RESULTS: The mean DD values of chitosan samples obtained by HBr titrimetry was significantly lower (p < 0.05) compared to IR spectroscopy using film (concentrations of 0.5 and 1 percent) as well as FDUV-spectrophotometry (purified and unpurified samples), but were significantly higher (p< 0.005) than those obtained using IR spectroscopy using KBr disk except for Chit-S2 computed using baseline (a), which were closely comparable (p > 0.05). In the IR spectroscopic method, the DD values differed when computed using different baseline. It can be observed that the values calculated using baseline (b) was generally higher than those calculated using baseline (a). On the other hand, chitosan samples prepared in the form of film might not be significantly affected by the different baseline as no consistent trend was observed. For FDUV-spectrophotometry, the DD values of purified Chit-S2 samples dried using oven and freeze dryer were comparable and not significantly different (p > 0.05). Similarly, no significant difference was observed for unpurified chitosan samples when dried using freeze dryer and oven (p > 0.05). Furthermore, the DD values of the purified and unpurified chitosan samples dried using freeze dryer were not significantly different except for Chit-S2. CONCLUSION: The DD values of chitosan were highly affected by the analytical methods employed. Hence, we proposed that the quantification method for DD should also be stated when reporting the DD value of chitosan sample.
Introduction
Chitosan is a natural polysaccharide comprising copolymers of glucosamine and N-acetylglucosamine, and can be obtained by the partial deacetylation of chitin, from crustacean shells, the second most abundant natural polymer after cellulose (1, 2). Chitosan has been widely used in vastly diverse fields, ranging from waste management to food processing, medicine and biotechnology (3). It becomes an interesting material in pharmaceutical applications (1) due to its biodegradability and biocompatibility (4), and low toxicity (5). Chitosan has found wide applicability in conventional pharmaceutical devices as a potential formulation excipient (6). The use of chitosan in novel drug delivery as mucoadhesive (7), peptide (8) and gene delivery (9), as well as oral enhancer (10) have been reported in the literature. Chitosan exhibits myriad biological actions such as hypocholesterolemic, antimicrobial, and wound healing properties (6, 11). Since chitosan is a new substance, it is important to carry out precise standardization for its pharmaceutical and biomedical applications like other auxiliary substances (12). Chitosan can be characterized in terms of its quality, intrinsic properties (purity, molecular weight, viscosity, and degree of deacetylation) and physical forms (13). Furthermore, the quality and properties of chitosan product may vary widely because many factors in the manufacturing process can influence the characteristics of the final product (14). Chitosan is commercially available from a number of suppliers in various grades of purity (7), molecular weight, and degree of deacetylation (15). It was reported that the degree of deacetylation is one of the more important chemical characteristics (14), which could influence the performance of chitosan in many of its applications (16, 17). In addition, the degree of deacetylation, which determines the content of free amino groups in the polysaccharides (14), can be employed to differentiate between chitin and chitosan. For instance, chitin with a degree of deacetylation of 75% or above is generally known as chitosan (18). The process of deacetylation involves the removal of acetyl groups from the molecular chain of chitin, leaving behind a complete amino group (-NH2 ) and chitosan versatility depends mainly on this high degree chemical reactive amino groups. There are methods available to increase or decrease the degree of deacetylation. For example, increase either in temperature or strength of sodium hydroxide solution could enhance the removal of acetyl groups from chitin, resulting in a range of chitosan molecules with different properties and hence its applications (17, 19). Since the degree of deacetylation depended mainly on the method of purification and reaction conditions (17, 18), it is therefore essential to characterize chitosan by determining its degree of deacetylation prior to its utilization at the developmental stage of drug delivery systems.
Various methods have been reported for the determination of the degree of deacetylation of chitosan. These methods included ninhydrin test (20), linear potentiometric titration (21), near-infrared spectroscopy (22), nuclear magnetic resonance spectroscopy (23), hydrogen bromide titrimetry (17, 24), infrared spectroscopy (24-26), and first derivative UV-spectrophotometry (27, 28). Some of the methods are either too tedious, costly for routine analysis (nuclear magnetic resonance spectroscopy), or destructive to the sample (Ninhydrin Test). Furthermore, many limit the range of degree of deacetylation to which they are applicable (24). Lately, the first derivative UV-spectrophotometry was advocated for the degree of deacetylation determination (27, 28). From the literature, the DD values of chitosan appeared to be highly associated with the analytical methods employed. A comprehensive study to examine three commonly used analytical methods, first derivative UV-spectrophotometry (FDUV-spectrophotometry), hydrogen bromide titrimetry (HBr) and infrared spectroscopy (IR) in the determination of chitosan degree of deacetylation have not yet been reported. Hence, the aim of the present study was to investigate the influence of the analytical methods on chitosan degree of deacetylation (DD) value.
Materials and Methods
Materials
Three commercially available chitosan samples were used. Two chitosan samples, Chit-S1, Lot #83H0036, practical grade from Crab Shells and Chit-S2, Lot #116H1465, from Crab Shells, minimum 85% deacetylated, were purchased from Sigma Chemical, St. Louis, USA, while another chitosan sample, Chit-F, Code #22743, high molecular weight, was obtained from Fluka Biochemica, Buchs, Switzerland. Glacial acetic acid, hydrobromic acid (47-50%) and sodium tetraborate were purchased from Sigma Chemical, St. Louis, USA. D-glucosamine hydrochloride and N-acetylglucosamine were purchased from Fluka, Buchs, Switzerland. Sodium hydroxide was purchased from R&M chemical, Essex, U.K. Ammonia solution 33% was obtained from May and Baker, England. Methanol GR was purchased from Merck, Darmstadt, Germany. All other reagents and solvents used were of analytical reagent grade. The materials were used as received.
Hydrogen Bromide Titrimetric Analysis
The method proposed by Sabnis and Block (29) was employed after slight modification. 0.5 g of chitosan was dissolved in 100 ml of freshly prepared 0.2 M hydrobromic acid. Concentrated 9M hydrobromic acid (50 ml) was then added to the chitosan solution with vigorous stirring to precipitate the hydrobromide salt. The resultant slurry was centrifuged at 2000 rpm for 30 min and the supernatant was discarded. The chitosan hydrobromide salt was then filtered off and washed several times with a mixture of methanol and diethyl ether (1:1, v/v) until the filtrate was neutral to litmus. Residual moisture in the chitosan hydrobromide salt was removed by stirring for six hours in anhydrous diethyl ether. After final filtration, the precipitate was dried in a vacuum desiccator for 12 hr to yield a white chitosan hydrobromide salt.
An accurately weighed (approximately 0.2 g) chitosan hydrobromide salt was dissolved in 100 ml of distilled water. The resulting solution (20 ml) was titrated against a standardised 0.1 M sodium hydroxide solution using phenolphthalein as an indicator. The moles of neutralized alkali corresponded to the moles of hydrobromic acid present, which corresponded to the mole of glucosamine units of chitosan initially present in the solution, thus facilitating the calculation of the degree of deacetylation of chitosan samples.
Infrared Spectroscopic Analysis
Chitosan samples prepared in the forms of potassium bromide (KBr) disk and film were studied. The KBr disk was prepared according to the method of Sabnis and Block (29) with slight modifications. Approximately 40-60 mg of chitosan powder and 120 mg of KBr was blended and triturated with agate mortar and pestle for 10 min. Approximately 40 mg of the mixture were compacted using a IR hydraulic press at a pressure of 8 tons for 60 s. The disk was conditioned in a desiccator placed in an oven at 80°C for 16 hr before analysis.
The chitosan films were prepared according to the method mentioned by Baxter et al. (17) with slight modifications. Chitosan films were prepared by casting 0.5 and 1.0% w/v chitosan in 1% acetic acid solutions, followed by drying in an oven at 60°C for 12 hr. The chitosan films were deprotonated by washing 3-4 times with methanolic ammonia (30) followed by distilled water and lastly methanol. The chitosan films were kept in a desiccator placed in an oven at 80°C for 16 hr before scanning.
The spectra of chitosan samples (in the forms of KBr disk and film) were obtained using an I.R. Instrument (MB-100, Bomem Hartmann & Braun, Quebec, Canada) with a frequency range of 4000-400 cm -1. The degree of deacetylation (DD) of the chitosan samples was calculated using two different baselines, baseline (a), which was proposed by Domszy and Roberts (24) and baseline (b) by Baxter et al. (17). The computation equations for the two baselines are given below:
DD = 100 - [(A1655 /A3450) X 100 / 1.33] -- Baseline (a)
where A 1655 and A 3450 were the absorbance at 1655 cm -1 of the amide-I band as a measure of the N-acetyl group content and 3450 cm -1 of the hydroxyl band as an internal standard to correct for film thickness or for differences in chitosan concentration powder form. The factor `1.33' denoted the value of the ratio of A 1655 /A 3450 for fully N-acetylated chitosan. It was assumed that the value of this ratio was zero for fully deacetylated chitosan and there was a rectilinear relationship between the N-acetyl group content and the absorbance of the amide-I band (24).
DD = 100 - [(A1655 / A3450) X 115] -- Baseline (b)
The baseline (b) proposed by Baxter et al. (17) was modified from the method reported by Domszy and Roberts (24).
The amide and hydroxyl bands absorbances can further be represented by the simple mathematical expressions as proposed by Sabnis and Block (29).
Log10 (DF1/DE) = (A1655)amide or Log10 (DF2/DE) = (A1655)amide
and
Log10 (AC/AB) = (A3450)hydroxyl
where DF 1 (for the base line `a') or DF 2 (for the base line `b'), DE, AC, and AB depicted the absolute heights of the absorption bands of the functional groups at their respective wavelengths. The absorbance ratio was calculated as follows:
Absorbance ratio = (A1655)amide / (A3450)hydroxyl
First Derivative UV-Spectrophotometric Analysis
The method reported by Tan et al. (28) was used after some modifications. A series of N-acetylglucosamine standard solutions of 0.005 - 0.050 mg/ml in 0.01 M acetic acid solution were prepared and their first derivative spectra recorded. The first derivative spectra of acetic acid solutions at concentrations of 0.01, 0.02 and 0.03 M was obtained using a UV-VIS spectrophotometer (Shimadzu 2100, Japan) in the range of 250-190 nm. The zero crossing point (ZCP) was determined by superimposing the spectra of these solutions. The vertical distance from ZCP to each N-acetylglucosamine solution spectrum, `H' was measured. A linear calibration curve was obtained by plotting the H values against the corresponding N-acetylglucosamine concentration.
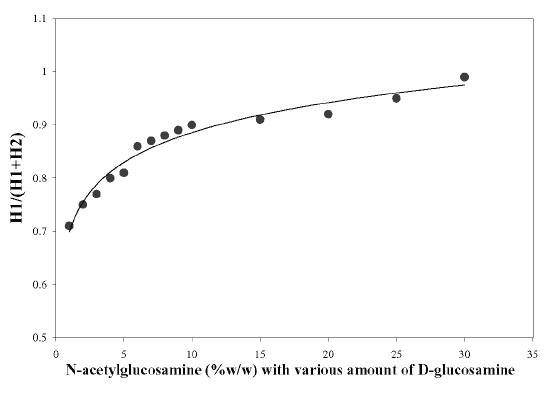
As the presence of glucosamine could give rise to a larger `H' value for N-acetylglucosamine, a reference curve was constructed for the correction of this effect (27). A 0.1 mg of N-acetylglucosamine (GlcNAc) per ml of 0.01 M acetic acid solution was prepared. On the other hand, N-acetylglucosamine (%w/w) of different concentrations was prepared by mixing different ratios of D-glucosamine (GlcN) and GlcNAc solutions. The peak height (H-value in mm) of the pure GlcNAc solution (H 1 ) and the solutions of different percentages of GlcNAc (H 2 ) was measured from the zero crossing point. Correction factors can be derived from the reference curve, which was obtained by plotting the H 1 /(H 1 + H 2 ) versus the corresponding N-acetylglucosamine concentrations (% w/w).
Prior to analysis, the chitosan samples were divided into two portions. One portion was analyzed as received (unpurified) while the other portion was further purified using the method reported by Tan et al. (28). The chitosan samples were further subdivided into two parts; one part dried using an oven at 80°C for 24 hr while the other using freeze dryer for 24 hr.
About 0.01 g of chitosan samples were dissolved in 10 ml of 0.01 M acetic acid solution and diluted up to 100 ml with distilled water. The first derivative spectrum of the sample was recorded. The vertical distance, H values (mm), was measured from ZCP and the contribution due to each N-acetylglucosamine was obtained from the calibration curve. The DD% of the unknown chitosan samples was determined using the following equation:
DD = 100 - [A / (W-204A) / 161+ A] X 100
where `A' is the amount of N-acetylglucosamine determined / 204 and `W' is the mass of chitosan used.
Statistical Analysis
The results were presented as mean ± standard deviation. The DD values obtained from the different methods were compared using a one-way analysis of variance. When a statistically significant difference was obtained (p<0.05), a Tukey-HSD test was then performed. On the other hand, the DD values obtained using two different baselines of IR spectroscopic analysis were compared using Independent Sample Student's t test.
RESULTS
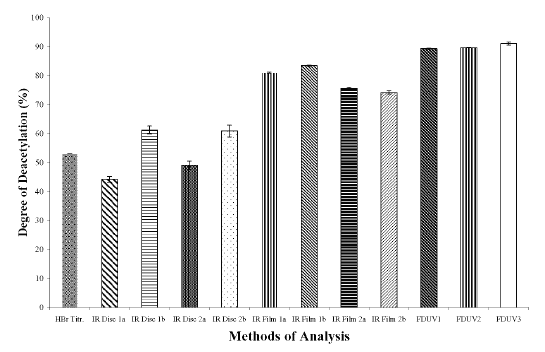
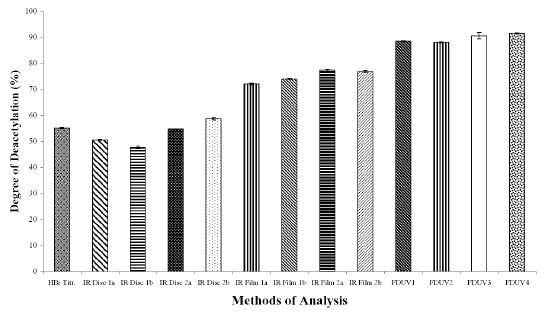
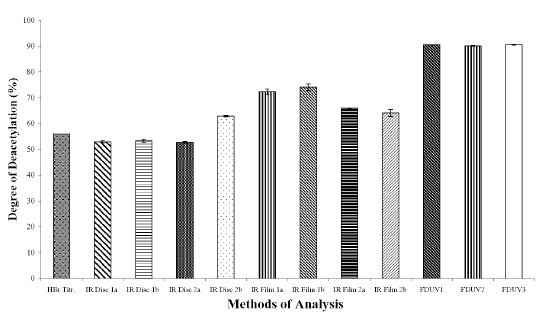
The DD values calculated using different analytical methods for the three chitosan samples (Figures 1A, 1B, and 1C) were significantly different (p < 0.05) for all the three chitosan samples, indicating that the DD values are highly dependent on the type of analytical methods employed. The DD values were noted to be the highest when calculated using FDUV-spectrophotometry, followed by IR spectroscopy using film, HBr titrimetry, and lastly IR spectroscopy using KBr disk.
Figure 1A: Degree of Deacetylation of Chitosan Sample (Chit-S1) Measured by Hydrogen BromideTitrimetry, Infrared Spectroscopy and First Derivative UV-Spectrophotometry. Mean ± SD, N = 3.
Figure 1B: Degree of Deacetylation of Chitosan Sample (Chit-S2) Measured by Hydrogen Bromide Titrimetry, Infrared Spectroscopy and First Derivative UV-Spectrophotometry. Mean ± SD, N = 3.
Figure 1C: Degree of Deacetylation of Chitosan Sample (Chit-F) Measured by Hydrogen Bromide Titrimetry, Infrared Spectroscopy and First Derivative UV-Spectrophotometry. Mean ± SD, N = 3.
The mean DD values of chitosan samples obtained by HBr titrimetry was significantly lower (p < 0.05) compared to IR spectroscopy using film (concentrations of 0.5 and 1%) as well as FDUV-spectrophotometry (purified and unpurified samples), but were significantly higher (p< 0.05) than those obtained using IR spectroscopy using KBr disk except for Chit-S2 computed using baseline (a), which were closely comparable (p > 0.05).
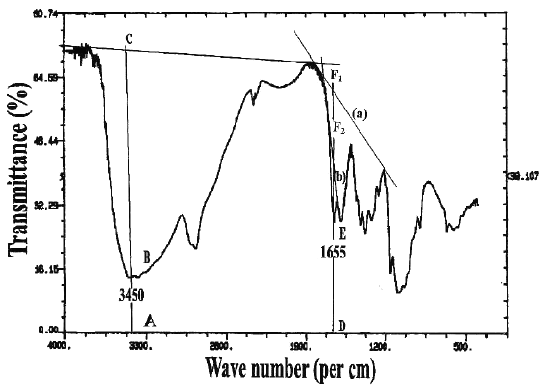
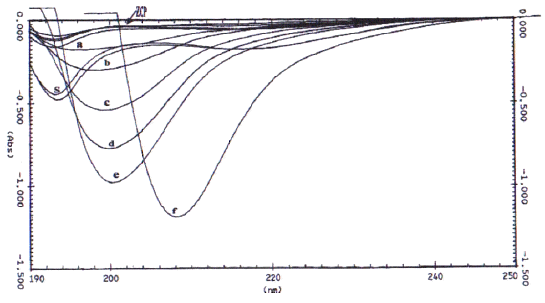
In the IR spectroscopic method, two different baselines were employed to analyze the DD values of chitosan samples, which were prepared in two different physical forms. The IR spectra with the respective baselines are illustrated in Figure 2.
Figure 2: I.R. spectrum of chitosan showing the two baselines (`a' and `b') for calculating the amide I band absorbance for the ratio A 1655 /A 3450 .
For chitosan samples prepared in the form of KBr disk, at ratios of 1:2 and 1:3, the DD values of Chit-S1, Chit-S2, and Chit-F, between the two different baselines were significantly different (p < 0.05), except at ratio 1:2 for Chit-F (p > 0.05). These results showed that DD values differ when computed using different baseline. It can be observed that the values calculated using baseline (b) was generally higher that those calculated using baseline (a). On the other hand, for chitosan in the form of film, the DD values were significantly different (p < 0.05) between baseline (a) and (b) at both concentrations for Chit-S1 and at 0.5% for Chit-S2, but not at 1% for Chit-S2 and at both concentrations for Chit-F. Chitosan samples prepared in the form of film might not be significantly affected by the different baseline as no consistent trend was observed.
From the Tukey-HSD test, the mean DD values of KBr at ratio 1:2, KBr at ratio 1:3, film at 0.5%, and film at 1% calculated using baseline (a), were found to be significantly different except between KBr at ratio 1:2 and KBr at ratio 1:3 for Chit-F (p > 0.05). As for baseline (b), all were found significantly different statistically (p < 0.05) except between KBr at ratio 1:2 and KBr at ratio 1:3 for Chit-S1 (p > 0.05) as well as between KBr at ratio 1:3 and film at 1% for Chit-F (p > 0.05).
Figure 3 shows the first derivative spectra of acetic acid solutions (0.01, 0.02 and 0.03 M), N-acetylglucosamine standards (0.005-0.050 mg/ml) and chitosan samples.
Figure 3: First derivative UV-spectra of various standard solutions of N-acetylglucosamine (a = 0.005, b = 0.01, c = 0.02, d = 0.03, e = 0.04, and f = 0.05 mg/ml in 0.01 M acetic acid), sample of chitosan (S), and zero crossing point (ZCP).
When the first derivative spectra of three different concentrations of the acetic acid solutions were recorded against water, it was noted that all the acetic acid spectra shared a common point at a wavelength of about 202 nm which was denoted as zero crossing point (28). Superimposition of these three spectra permitted the exact individualization of the zero crossing point (ZCP) of the acid at 202 nm. The ZCP corresponded to the N-acetylglucosamine maximum on the wavelength axis, and this makes the N-acetylglucosamine determination independent of the acetic acid concentration range usually encountered with dilute chitosan solutions (31).
The correlation coefficient (r 2 ) between `H' values and concentrations of N-acetylglucosamine (GlcNAc) was calculated to be 0.998. The first derivative spectra of unknown chitosan samples (S in Figure 3) were recorded. The `H' (mm) values from ZCP were measured and the contribution due to GlcNAc was obtained from the calibration curve. The experimental data showed that the presence of glucosamine (GlcN) could deliver a larger `H' values for N-acetylglucosamine solutions than expected. Muzzarelli and Rochetti (27) reported that the presence of GlcN contributed to the `H' value when the GlcNAc content was less than 10%. On the other hand, the results produced by Tan et al. (28) demonstrated that this effect was prevalent when the GlcNAc was 20%. In view of these discrepancies, a reference curve was constructed in the present study as shown in Figure 4. It was observed that the contribution of glucosamine to the `H' value was prominent even when the GlcNAc content was at 30%.
Figure 4: Correction curve for the determination of N-acetylglucosamine.
DISCUSSIONS
The variation in the values of DD among the three chitosan samples used in the study was anticipated as their manufacturing process and condition differed from one to another. The dependence of DD on the source and method of purification has been reported in the literature (17, 18). Other research groups also reported the dependency of DD values on the type of analytical methods used (17, 27). The varied values of DD for the same batch of chitosan could be ascribed to different sample preparation, experimental conditions as well as type of instrument used and its sensitivity (29, 32). The HBr titrimetric method has the advantage that it measured the protonated amine groups directly. Furthermore, there was no problem of lack of accessibility of the amine groups during the protonation step as the salt was formed by precipitation from solution using acid (24). Since the chemical basis of this method is based on reaction with the amine group, the presence of protein contaminants remained in the sample during the extraction process could adversely interfere the results. During the recovery of chitosan HBr salt, the filtrate was washed several times with a mixture of methanol and diethyl ether until neutral to litmus. The use of litmus paper could not indicate accurately the actual neutral pH value, leading to a false reflection of a complete removal of HBr. As a result, the chitosan HBr salt might not be completely free from HBr, which would ultimately affect the DD values of chitosan. In addition, the use of phenolphthalein as an indicator of the end point in the titration of the amino group could also result in lower DD values.
IR spectroscopic method, which was initially proposed by Moore and Roberts (25), is commonly used for the estimation of chitosan DD values. It has a number of advantages because it is relatively fast and does not require dissolution of the chitosan sample in an aqueous solvent (17, 29). Nevertheless, IR spectroscopy is primarily a solid-state method utilizing baseline for DD calculation. Employment of different baseline would inevitably contribute to variations in the DD values (17, 26). Furthermore, sample preparation, type of instrument used and conditions may influence the sample analysis (26, 29). In addition, there may be possible argument for choosing A3450 absorption band as an internal standard as errors may arise due to the effect of adsorbed water on the intensity of the hydroxyl band (24). Chitosan is hygroscopic in nature and it was reported that the capability of moisture adsorption of chitosan decreased with an increase in deacetylation (32). This suggests that samples having higher DD may absorb less moisture than those with lower DD. Moreover, experimental errors may occur during weighing of the samples. As such, it is essential that the samples under observation should be completely dry (32). In the present study, this was circumvented by conditioning the chitosan samples in a desiccator placed in a circulating air oven at 80°C for 16 hr. In the case of films, it was washed for 3-4 times with methanolic ammonia for deprotonation, followed by washing with distilled water and finally by methanol. The deprotonation process was anticipated to be complete as this could adversely affect the DD values. As such, it can be concluded that different physical forms contribute to different DD values.
In the FDUV-spectrophotometric method, comparisons using Tukey-HSD test showed that the DD values of purified Chit-S2 samples were comparable and not significantly different (p > 0.05) when dried in oven and freeze dryer, indicating that the sample can be dried using an oven, which provides a more economical means of drying and hence can reduce the overall cost of analysis. Tan et al. (28) purified the chitosan samples to remove the impurities followed by freeze-drying. Since freeze-drying is an expensive process, we explored an alternative means of drying for chitosan samples using oven. Similarly, no significant difference was observed for unpurified chitosan samples when dried using freeze dryer and oven (p > 0.05). Furthermore, the DD values of the purified and unpurified chitosan samples dried using freeze dryer were not significantly different except for Chit-S2. Hence, it can be suggested that the chitosan samples used in the present study could be of high purity and therefore may not require purification.
The FDUV-spectrophotometric method requires only a small amount of sample and relies on simple reagents and can be analyzed by common laboratory spectrophotometer. It permits a simple, convenient, time saving and is sensitive enough to detect the concentration of GlcNAc as low as 0.0005 mg/ml. in 0.01 M acetic acid. The results obtained are quite reasonable with less interference of protein contaminants, which provides a good precision and accuracy for the assay of N-acetylglucosamine residues in chitosan. The use of water as a reference blank avoids light absorption in the optical system thus permitting a better signal to noise ratio. On the other hand, the setback of using FDUV-spectrophotometric method for the determination of DD of chitosan was that the results which depended mainly on H-values (mm) were obtained manually, which appeared to be crude and depended highly on operator.
CONCLUSIONS
It can be concluded that the DD values were highly affected by the analytical methods employed. Hence, we proposed that the analytical method used for the quantification of the DD values should be stated when reporting DD values of chitosan products.
References
Illum, L., Chitosan and Its use as a Pharmaceutical Excipient. Pharm Res., 15:1326-1331, 1998.
Nunthanid, J., Puttipipatkhachorn, S., Yamamoto, K. and Peck, G.E., Physical Properties and Molecular Behavior of Chitosan Films. Drug Dev Ind Pharm., 27 (2): 143-157, 2001.
Savant, V. and Torres J.A., American Chitoscience Society 1: 1-4, 1995.
Borchard, G and Junginger H.E., Modern drug delivery applications of chitosan. Adv Drug Del Rev., 52 (2): 103, 2001.
Karlsen, J. and Skaugrud, O., Excipient Properties of chitosan. Manuf Chem., 62: 18-19, 1991.
Singla, A.K. and Chawla, M., Chitosan: some pharmaceutical and biological aspects-an update. J Pharm Pharmacol., 53 (8): 1047-1067, 2001.
Lehr, C.M., Bouwstra, J.A., Schacht, E.H. and Junginger, H.E. In vitro evaluation of mucoadhesive properties of chitosan and some other natural polymers. Int J Pharm., 78: 43-48, 1992.
Bernkop-Schnurch, A. and Kast, C.E., Chemically modified chitosans as enzyme inhibitors. Adv Drug Del Rev., 52 (2): 127-137, 2001.
Borchard G., Chitosans for gene delivery. Adv Drug Del Rev., 52 (2): 145-150, 2001.
Thanou, M., Verhoef, J.C. and Junginger, H.E., Oral drug absorption enhancement by chitosan and its derivatives. Adv Drug Del Rev., 52 (2): 117-126, 2001.
Ueno, H., Mori, T. and Fujinaga, T., Topical formulations and wound healing applications of chitosan. Adv Drug Del Rev., 52 (2): 105-115, 2001.
Knapczyk, J., Krowczynski, L., Krzek, J., Brzeski, M., Murnberg, E., Schenk, D. and Struszczyk, H., Requirement of chitosan for pharmaceutical and biomedical application, in Skjak-Braek, G., Anthonsen, T. and Sanford, P., (eds), Chitin and Chitosan-Sources, Chemistry, Biochemistry, Physical Properties and Applications, Elsevier, London, pp 657- 663, 1989.
Sanford, P.A., Chitosan: commercial uses and potential applications, in Skjak-Braek, G., Anthonsen, T. and Sanford, P., (eds), Chitin and Chitosan-Sources, Chemistry, Biochemistry, Physical Properties and Applications, Elsevier, London, pp 51-70, 1989.
Li, Q., Dunn, E.T., Grandmaison E.W. and Goosen, M.F.A., Applications and Properties of Chitosan. J Bioact Compat Polym., 7: 370-397, 1992.
Genta, I., Perugini, P. and Pavanetto, F., Different molecular weight chitosan microspheres: Influence on drug loading and drug release. Drug Dev Ind Pharmacy., 24:779-784, 1998.
Muzzarelli, R.A.A, Chitin, Pergamon Press, Oxford, 1977.
Baxter, A., Dillon. M., Taylor. K.D.A. and Roberts. G.A.F., Improved method for i.r. determination of the degree of N-acetylation of chitosan. Intl J Biol Macromol., 14: 166-169, 1992.
Li, J., Revol, J.F. and Marchessault, R.H., Effect of degree of Deacetylation of Chitin on the Properties of Chitin Crystallites. J Appl Polym Sci., 65(2): 373-380, 1997.
Mima, S., Miya, M., Iwamoto, R. and Yoshikawa, S., Highly Deacetylated Chitosan and Its Properties. J Appl Polym Sci., 28(6): 1909-1917, 1983.
Curotto, E. and Aros, F., Quantitative determination of chitosan and the percentage of free amino groups. Anal Biochem., 211:240-241, 1993.
Ke, H. and Chen, Q., Potentiometric titration of chitosan by linear method. Huaxue Tongbao., 10:44-46, 1990.
Rathke, T.D. and Hudson, S.M., Determination of the Degree of N-Deacetylation in Chitin and Chitosan as well as Their Monomer Sugar Ratios by Near Infrared Spectroscopy. J Polym Sci: Part A: Polym Chem., 31:749-753, 1993.
Hiral, A., Odani, A. and Nakajima, A., Determination of degree of deacetylation of chitosan by 1H NMR spectroscopy. Polym Bull., 26:87-94, 1991.
Domszy, J.G. and Roberts, G.A.F., Evaluation of infrared spectroscopic techniques for analyzing chitosan. Makromol Chem., 186:1671-1677, 1985.
Moore, G.K. and Roberts, G.A.F., in Muzzarelli, R.A.A. and Pariser, E.R., (eds), Proceedings of the first International Conference on chitin/chitosan. MIT Sea Grant Report 78-7, pp 421, 1978.
Sannan, T., Kurita, K., Ogura, K. and Iwakura, Y., Studies on chitin: 7. I.r. spectroscopic determination of degree of deacetylation. Polym., 19:458-459, 1978.
Muzzarelli, R.A.A. and Rocchetti, R., Determination of the Degree of Acetylation of Chitosans by First Derivative Ultraviolet Spectrophotometry. Carbohydr Polym., 5: 461-472, 1985.
Tan, S.C., Khor, E., Tan, T.K. and Wong, S.M., The degree of deacetylation of chitosan: advocating the first derivative UV-spectrophotometry method of determination. Talanta, 45:713-719, 1998.
Sabnis, S. and Block, L. H., Improved infrared spectroscopic method for the analysis of degree of N-deacetylation of chitosan. Polym Bull., 39:67-71, 1997.
British Pharmacopoeia. British Pharmacopoeia Commission Her Majestys Stationery Office, Norwich, London. Vol. II, Appendix V F. A 144, 1999.
Muzzarelli, R.A.A., Rochetti, R., Stanic, V. and Weckx, M., Methods for the determination of the degree of acetylation of chitin and chitosan, in Muzzarelli, R.A.A. and Peter, M.G., (eds), Chitin Handbook, European Chitin Society, Ancona, Italy, pp 109-119, 1997.
Blair, H.S., Guthrie, J., Law, T.K. and Turkington, P., Chitosan and Modified Chitosan Membranes I. Preparation and Characterization. J Appl Polym Sci., 33:641-656, 1987.
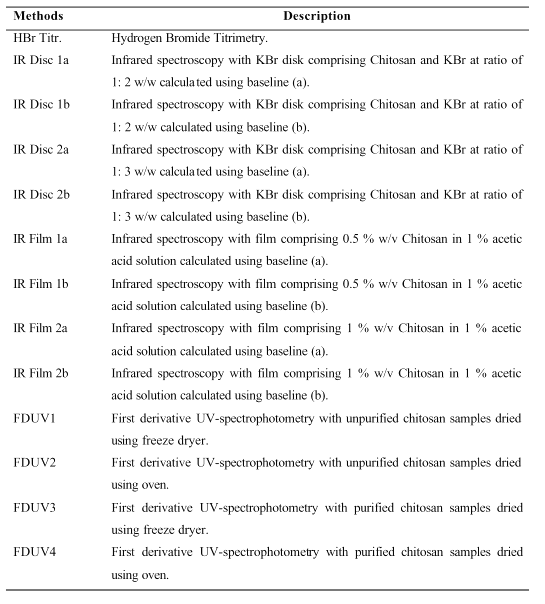
Table 1: List of abbreviations used for DD Methods of analysis in Figures 1A, 1B and 1C.
Corresponding Author: Kok Khiang Peh, School of Pharmaceutical Sciences, University of Science Malaysia, 11800 Penang, Malaysia, kkpeh@usm.my
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps