J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 5(3):245-257, 2002
Molecular modeling of potential new and selective PET radiotracers for the serotonin transporter.
Julia Wellsow, Karl-Artur Kovar1
Pharmaceutical Institute, Department of Pharmaceutical and Analytical Chemistry, University of Tübingen, Auf der Morgenstelle, Tübingen, GermanyHans-Jürgen Machulla
Department of Radiopharmacy / PET Center, University of Tübingen, Röntgenweg, Tübingen, GermanyReceived 27 August 2002, Revised 9 October 2002, Accepted 10 October 2002
Abstract
PURPOSE: Imaging the serotonin transporter (SERT) with Positron Emission Tomography (PET) provides a useful tool for understanding alterations of the serotonergic system. However, no optimal PET radiotracer for the SERT yet exists. The main purpose of this study was to design potential new and selective PET radiotracers for the SERT and to predict their binding affinity at both the SERT and the norepinephrine transporter. METHODS: Molecular Modeling was used for ligand design. Predictions of binding affinity were based on models generated by Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA). RESULTS: A series of 100 compounds were suggested. As diphenyl sulfide derivatives like [11C] DASB have recently proven to be promising PET ligands, rational modification of the diphenyl sulfide scaffold has been performed. The novel compounds were predicted to be selective high affinity SERT ligands. Important new ideas are the introduction of a fluoroethyl-oxycarbonyl group (ester) and a fluorethyl-carbonyl group (ketone), as well as a formyl group (aldehyde), and its corresponding oxime and imine. Another innovative suggestion is the replacement of the sulfur bridge with a cyanamide group and a fluoroethylamino group. CONCLUSIONS: The suggested compounds possess features providing new possibilities for carbon-11 or fluorine-18 labeling. Synthesis, biological testing, and screening for PET suitability are reasonable further steps.
Introduction
The serotonergic neurotransmission plays an important role in the central nervous system. Alterations of serotonin (5-HT) levels are associated with many psychiatric disorders. The serotonin transporter (SERT), located on presynaptic nerve endings, modulates synaptic 5-HT levels. It also functions as the primary target site for many antidepressant drugs. Moreover, drugs of misuse like 3, 4-methylenedioxymethylamphetamine (MDMA, "ecstasy") and to some extent cocaine are known to exert their effect via the 5-HT reuptake site. For these reasons, in vivo mapping of the SERT in the living human brain by positron emission tomography (PET) is most valuable for understanding alterations of the serotonergic system and might prove useful in monitoring antidepressant therapy. However, PET investigations of the SERT have been limited by the small number of candidate radioligands and their various shortcomings. Therefore, there is considerable interest in the development of a suitable PET radioligand for the SERT.
Necessary requirements for a successful PET radioligand are not only a high binding affinity at the target site, but also a high selectivity, rapid crossing of the blood brain barrier, a high specific to non-specific binding ratio, suitable brain kinetics, and good synthetic availability. PET radioligands are usually labeled with either carbon-11 or fluorine-18. This is preferably done in the last synthesis step. Replacement of carbon-12 by carbon-11 results in compounds that are physiologically indistinguishable from their unlabeled counterparts. Replacement of a hydrogen atom or hydroxyl group with fluorine also very often retains or even enhances the biological activity of a molecule (1). Carbon-11 has a half-life of 20 min, whereas fluorine-18 has a half-life of 110 min. This longer half-life of fluorine-18 can be advantageous when using PET ligands with slow kinetics. Several classes of compounds have been screened for their suitability as PET ligands for the SERT.
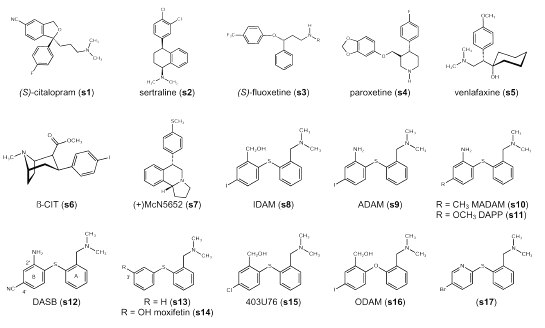
Figure 1: Molecular structures.
Scheffel et al. (2) give a comprehensive overview of early investigations. Oh et al. (3) and Laakso et al. (4) summarize recent developments. Selective serotonin reuptake inhibitors (SSRIs) such as citalopram (s1), sertraline (s2) and fluoxetine (s3) (Figure 1) have been labeled with carbon-11, but a relatively poor signal-to-noise ratio limited their use in vivo for the quantification of the SERT (2, 5-7). Carbon-11 labeled venlafaxine (s5) also turned out not to be an ideal PET ligand (8). Neither carbon-11 labeling nor fluorine-18 labeling of paroxetine (s4), another SSRI with high affinity for the SERT, has been accomplished yet (2). Many tropane and nortropane analogs have been investigated as PET ligands for the SERT, such as [11C]b-CIT (2-b-carbomethoxy-3-b-(4-iodophenyl)-tropane) (s6) and [11C]nor-b-CIT (2-b-carbomethoxy-3-b-(4-iodophenyl)-nortropane) (9, 10). However, one major drawback is their lack of selectivity for the SERT over the dopamine transporter (DAT). Some new derivatives like ZIENT (2-b-carbomethoxy-3-b-(4-((Z)-2-iodoethenyl)phenyl)-nortropane) (11) and FEINT (2-b-carbomethoxy-3-b-(4-(2-fluoroethyl)-3-iodophenyl)-nortropane) (12) display high specific binding at the SERT and have been proposed as potential PET ligands. The most widely used radiotracer to date for PET imaging of the SERT is [11C](+)McN5652 (trans-1,2,3,5,6,10b-hexahydro-6-[4-(methylthio)phenyl]-pyrrolo[2,1-a]isoquinolone]) (s7) (13, 14), a potent blocker of 5-HT reuptake. McCann et al. (15, 16) could prove neurotoxic effects of MDMA on the serotonergic system in the human brain by using [11C] (+) McN5652 as a radiotracer. However, [11C](+)McN5652 also displays moderate affinity at the norepinephrine transporter (NET) and the DAT; and the low specific to non-specific binding ratio observed with [11C](+)McN5652 in humans limits its application as a PET imaging agent in vivo (14).
Only recently, several substituted diphenyl sulfides such as IDAM (5-iodo-2-((2-(dimethylaminomethyl)-phenylsulfanyl)benzylalcohol) (s8) (17), ADAM (N,N-dimethyl-2-(2-amino-4-iodophenylsulfanyl)benyzlamine) (s9) (18), MADAM (N,N-dimethyl-2-(2-amino-4-methylphenylsulfanyl)benzylamine) (s10) (19), DAPP (N,N-dimethyl-2-(2-amino-4-methoxyphenylsulfanyl)benzylamine) (s11) (20) and DASB (3-amino-4-((2-dimethylaminomethyl)-phenylsulfanyl)benzonitrile) (s12) (20, 21) have been described as potent and selective SERT ligands. These investigations were originally based on early studies that described moxifetin (s14) (22) and 403U76 (s15) (23) as novel antidepressants. Whereas iodine-123 labeled IDAM (s8) and ADAM (s9) have been described as suitable radioligands for in vivo visualization of the SERT using single photon emission computerized tomography (SPECT) in primates (17), carbon-11 labeled DAPP (s11) and DASB (s12) have shown favorable PET characteristics in the human brain (24). Also carbon-11 labeled MADAM (s10) might prove a suitable PET ligand for imaging the SERT (19, 25). These studies have shown that the diphenyl sulfides have promising characteristics for imaging the SERT. However, yet, no optimal PET ligand has been found among them. Slow kinetics preclude ADAM (s9), for instance, from being a useful PET tracer, and [11C]DASB (s12) is not appropriate to detect SERT in the cortex (26). Therefore, further structure-activity relationship (SAR) studies of the diphenyl sulfides are both necessary and warranted, particularly as the lack of an asymmetric center in these agents makes synthesis comparatively easy.
The aim of this study was to design potential new and selective PET radiotracers for the SERT by molecular modeling and to predict their binding affinity at both the SERT and the structurally similar NET. As the basis for design and prediction, models that quantify SARs of SERT ligands were to be used. Such Quantitative Structure-Activity Relationships (QSAR) models had earlier been developed in our group by Comparative Molecular Field Analysis (CoMFA) and by Comparative Molecular Similarity Indices Analysis (CoMSIA) (27) as no appropriate QSAR models could be found in literature. The diphenyl sulfide scaffold of the promising PET ligand DASB (s12) was to be used as lead for further structural modification. Possibilities for radiolabeling were to be considered when modifying the structures.
Methods
Computational approach
All molecular modeling studies were performed using SYBYL 6.6 (28) running on a Silicon Graphics Octane (R 10000) workstation.
Biological data
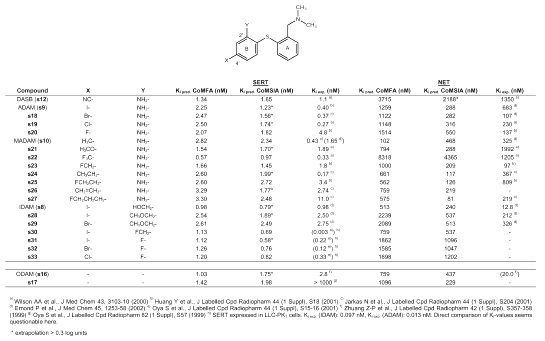
Binding affinity data of [11C]DASB (s12) and its derivatives at the SERT and the NET were taken from literature (21, 26, 29). Ki -values are given in Table 2.
Compound generation
DASB (s12) (Figure 2) was constructed using SYBYL´s fragment library. It was generated both in its N-protonated and non-protonated form. An initial geometry optimization was performed in vacuo using the Tripos Force Field (30) and the following non-default settings: Method : Conjugate Gradient, Termination Gradient : 0.01 kcal/mol*Å, Max. Iterations: 10,000. The resulting structure was used as starting geometry for the alignment and for a conformational search. The Genetic Algorithm Similarity Program (GASP) (31) served as alignment tool. Analogously to the alignment procedure used for model derivation (27) (S)-citalopram (s1) was used as template upon which DASB (s12) was superimposed together with (S)- fluoxetine (s3), paroxetine (s4) and sertraline (s2). Default settings were used and ten alignments were generated during each GASP run. The conformation used for the alignment (Figure 2) was chosen by visual inspection and was further minimized in vacuo using the Tripos Force Field (30) and the following non-default settings: Method : Conjugate Gradient, Termination Gradient : 0.01 kcal/mol*Å, Max. Iterations: 1000. To assure that the chosen conformation was a low-energy one, a Genetic Algorithm (GA) conformational search using default parameters was carried out. The conformation used for the alignment was found among the ten lowest-energy conformations. Assuming the same alignment for DASB (s12) and its derivatives, all DASB derivatives were constructed from their respective parent compound using SYBYL´s fragment library. They were minimized using the following non-default settings: Method : Conjugate Gradient, Termination Gradient : 0.01 kcal/mol*Å, Max. Iterations : 100. Partial atomic charges were calculated using the Gasteiger-Hueckel method.

Figure 2: Alignment of DASB and (S)-citalopram generated by using GASP.
CoMFA and CoMSIA
Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Indices Analysis (CoMSIA) are most often used in drug discovery to find the common features that are important in binding to the biologically relevant target. Both techniques are based on the assumption that changes in binding affinities of ligands are related to changes in different fields surrounding the molecules. These fields can be of steric and electrostatic, of hydrophobic, or of hydrogen-bond accepting and hydrogen-bond donating nature. Quantitative Structure-Activity Relationships (QSAR) are generated by multivariate statistics using Partial Least Squares (PLS) (32, 33) and can be applied for predicting the binding affinity of new molecules. CoMFA was introduced by Cramer et al. (34). CoMSIA is an extension of the CoMFA method recently developed by Klebe et al. (35, 36) and differs from CoMFA in the implementation of the fields.
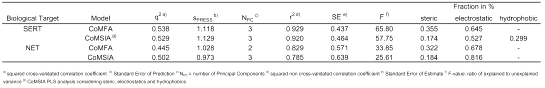
Table 1: CoMFA and CoMSIA analyses used for prediction.
Prediction of new substances and their binding affinities
The CoMFA and CoMSIA models established earlier in our group (27) served as starting-point for the virtual development of new substances. They were based on a diverse set of antidepressants (37). For the prediction of the binding affinities at the SERT and at the NET, we used the CoMFA PLS analysis considering sterics and electrostatics, and the CoMSIA PLS analysis considering sterics, electrostatics and hydrophobics (Table 1). These models were chosen as they predict the binding affinity of DASB (s12) both at the SERT and at the NET in the correct order of magnitude (Table 2).
Table 2: Predicted and experimental binding affinities of diphenyl sulfide derivatives.
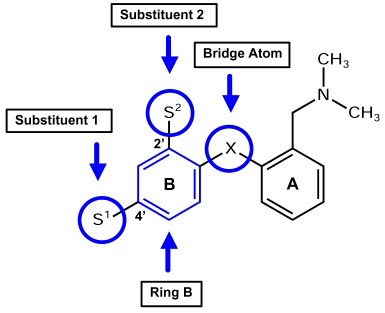
All CoMFA calculations were performed with the Tripos Advanced CoMFA Module (28). DASB (s12) served as lead structure. The Optimize QSAR interface was used to build a series of analogs and make a preliminary survey of their expected activities against a known QSAR (38). In this study, DASB´s (s12) core structure was provided as molecular scaffold, and S1 and S2 were specified as interchangeable substituents (Figure 3).
Figure 3: Positions of possible modifications at the diphenyl sulfide scaffold. Substituents in position 2' and 4' were exchanged, as well as the aromatic ring B, and the atom bridging ring A and ring B.
New structures are automatically suggested by screening a database of possible substituents. The following default settings were used: Configuration Options : Standard ( Conformational Refinement : All-trans, Computation of Charges: Gasteiger-Hueckel), Method : Random, Cycles: 100. The 12 best hits were retrieved. The CoMFA PLS analysis considering sterics and electrostatics was used (Table 1).
Predictions of target properties are most reliable if extrapolation with respect to topologies and functionalities does not occur, but small extrapolations of descriptors that make only a small contribution to the model are not a cause of particular concern. Extrapolation is described by the total contribution made to the prediction by the out-of-range descriptors (38). In predicting biological activity on a common log scale, any extrapolation below 0.3 log units is probably acceptable (38). This is the case for most CoMFA predictions. Some CoMSIA predictions, however, do not comply with this requirement and are indicated with an asterisk in Table 2 to 5.
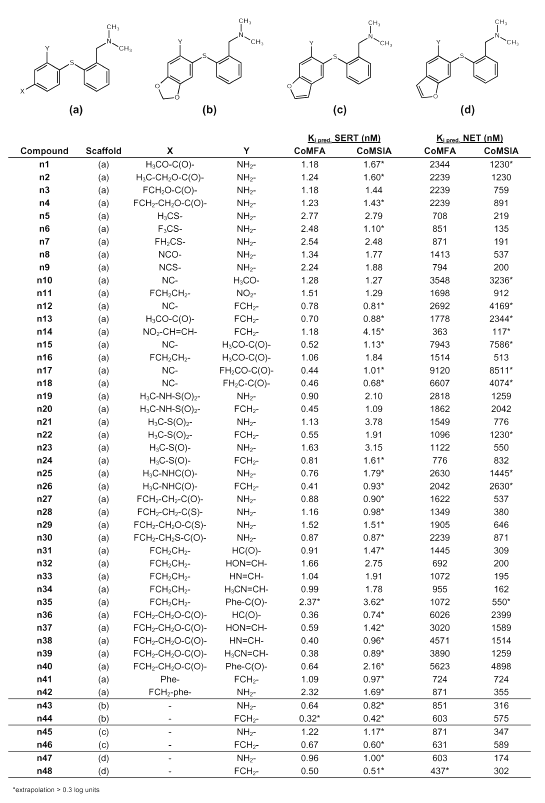
Table 3: Predicted binding affinities of novel diphenyl sulfides.
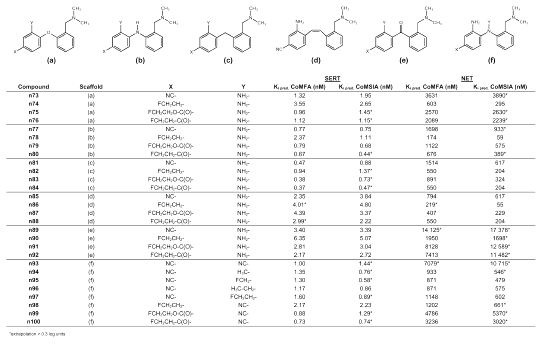
Table 4: Predicted binding affinities of novel phenylpyridinyl sulfides and phenyl thienyl sulfides.
Table 5: Predicted binding affinities of novel compounds with different diphenyl bridging.
Results And Discussion
General approach
As the diphenyl sulfides had turned out to be promising PET ligands for the SERT, the endeavor was to find even more suitable substances of this type. (Figure 3.) To structurally modify the lead compound DASB (s12), knowledge on structure-activity relationships (SARs) of SSRIs derived from our earlier developed CoMFA and CoMSIA models (27) was used. Moreover, known SARs for diphenyl sulfides were considered, as well as the concept of bioisosterism in drug design. Additionally, structures were automatically suggested by using SYBYL´s Optimize QSAR module (28).
Structure-activity relationships of diphenyl sulfides
Structure-activity relationships (SARs) of diphenyl sulfides were first described by Sindelar et al. (22, 39). Whereas structure (s13) (Figure 1) shows high binding affinity at both the SERT and the NET, moxifetin (s14), additionally possessing a hydroxy group in position 3´, is SERT selective (40, 41). Any further modifications at the moxifetin scaffold made by Sindelar et al. (22, 39) resulted in compounds possessing a substituent in position 3´. This changes with Ferris et al. (23) describing 403U76 (s15) as a new potential antidepressant inhibiting both the 5-HT and norepinephrine (NE) reuptake. The 2´, 4´-substitution pattern seen in 403U76 (s15) is maintained throughout further ligand optimization for SPECT and PET applications.
A comprehensive overview of the SARs of 2´, 4´-substituted diphenyl sulfides is given by Emond et al. (25). The dimethylaminomethyl group in position 2 of ring A seems to be optimal although a monomethylaminomethyl group is also tolerated. At position 2´ of ring B, the SERT binding site tolerates quite a large range of functional groups containing a heteroatom, such as alcohol, ether, ester, nitro and amine functions. The nature of this group seems to be important for SERT selectivity. Whereas compounds containing a hydroxymethyl group in position 2´ (IDAM (s8)) display high binding affinity at both the SERT and the NET, compounds substituted with an amino group (ADAM (s9)) or a methoxymethyl group (s28, s29) in position 2´ are SERT selective (25). Oya et al. (42) recently reported a new compound substituted with a fluorine-18 labeled fluoromethyl group in position 2´ (s30) displaying very high binding affinity at the SERT, and showing promising characteristics as a possible PET ligand. As no affinity data for the NET or the DAT is given, no conclusions about SERT selectivity can be drawn. Choi et al. (43) reported that a fluoro substitution is also tolerated in position 2´ (s31, s32, s33). It was further reported that arylation and acylation of the amino group in position 2´ in most cases resulted in reduced binding affinity at the SERT. Regarding the 4´ position of ring B, Emond et al. (25) concluded that the nature of the substituent only slightly influences the SERT binding affinity, as for instance a fluorine, a bromine and a iodine atom seem to be interchangeable in this position. This is supported by the findings of Wilson et al. (21) showing that also a chlorine atom (s19), a trifluoromethyl (s22), a methoxy (s21) and a cyano (s12) group in position 4´ display high SERT binding affinity. However, not all substituents are favorable as PET ligands. The trifluoromethyl group, for instance, is much too lipophilic, thus displaying a clearance from the cerebellum too slow for favorable pharmacokinetics in human PET studies (21). According to Emond et al. (25), sterically demanding groups in position 4´ markedly decrease binding affinity at the SERT. Yet Jarkas et al. (29) found that both an ethyl (s24) and an ethenyl (s26) group in position 4´ display a satisfying binding affinity at the SERT. Huang et al. (26) reported that a 2-fluoroethyl group in position 4´ (s25) also displays a good binding affinity at the SERT but additionally, a moderate binding affinity at the NET. Zhuang et al. (44) prepared an analog of IDAM (s8) called ODAM (s16) with an oxygen instead of a sulfur bridge between the two phenyl rings as this was thought to be more stable against metabolism. ODAM (s16) showed high binding affinity at the SERT and moderate binding affinity at both the NET and the DAT. However, Acton et al. (45) concluded that, although ODAM (s16) seems to have a higher brain uptake than IDAM (s8), and indeed a slower metabolism, it also exhibits higher nonspecific binding than IDAM (s8), which makes it less suitable as a PET ligand.
3D QSAR models
The outcome of our earlier 3D QSAR studies (27) is exemplified by the CoMFA model for the SERT. Figure 4 shows the steric (a) and electrostatic (b) contour diagram of the CoMFA model, displaying those features that are important for high binding affinity at the SERT.
Figure 4: CoMFA steric (a) and electrostatic (b) "StDev*Coeff" plots for the serotonin transporter. The contour levels were derived from examining the field value histograms. Green areas indicate regions where steric bulk favorably effects binding affinity. Yellow regions denote areas in which sterically demanding groups have a detrimental effect on binding affinity. Blue contours enclose areas where partial positive charges increase binding affinity whereas in the regions enclosed by red contours partial negative charges are favored. DASB (s12) is shown within the fields.
DASB (s12) is shown within the fields as an example for a SERT selective compound. The three green regions show those areas where steric bulk enhances affinity. These are the regions where the substituent in DASB´s (s12) position 4´ at ring B is located, the area around ring A and the area into which one of DASB´s (s12) N-methyl groups is orientated. The areas indicated by yellow contours should be sterically avoided; otherwise reduced affinity can be expected. A blue area, which represents a region where electron-deficiency is advantageous, can be seen around ring B. This can be attributed to ring substituents with a strong electron withdrawing effect such as the cyano group in DASB (s12). In regions enclosed by red areas more negative charges are favored. One red area is found between ring A and ring B; another red area is seen around DASB´s (s12) cyano group, showing that an electronegative substituent is favorable here. Analogous contour diagrams had been generated for the CoMSIA study at the SERT, for the CoMFA and the CoMSIA studies at the NET, and for selectivity analysis (27).
Comparison of predicted and experimental binding affinity
To check whether the CoMFA and CoMSIA models (27) are suitable to predict SERT binding affinity of new diphenyl sulfides, the equilibrium inhibition constants (Ki-values) of several diphenyl sulfides already synthesized and biologically tested were predicted. Predicted and experimental Ki-values for both the SERT and the NET are shown in Table 2. Clearly, one has to be careful with comparison of data across laboratories. However, the comparison of Ki-values seems permissible as the Ki-value is an absolute value for a compound, and is independent of the specific radioligand used and the concentration of radioligand in the assay (37). For DASB (s12), a Ki-value of 1.1 nM at the SERT and of 1350 nM at the NET was experimentally determined (21), showing that DASB (s12) is highly selective for the SERT. This is correctly predicted by the CoMFA and CoMSIA analyses, the predicted Ki-values being within the same order of magnitude as the experimentally determined ones with 1.34 nM (CoMFA) and 1.65 nM (CoMSIA) for the SERT, and 3715 nM (CoMFA) and 2188 nM (CoMSIA) for the NET. The Ki-values of compound s25, bearing a 2-fluoroethyl group in position 4´, are also predicted within the correct order of magnitude for both the SERT and the NET. This is of particular interest, as the 2-fluoroethly group is suitable for fluorine-18 labeling. This was shown only recently by Huang et al. (46) who prepared fluorine-18 labeled s25. However, judging from binding affinity data given in (26), compound s25 is less SERT selective than DASB (s12). This probably makes it less suitable as PET radiotracer, although Huang et al. (46) were recently able to demonstrate high specificity during PET studies in rats and baboons. The binding affinity of IDAM (s8), possessing a hydroxymethyl group in position 2´, and a iodine atom in position 4´, is correctly predicted as being in the sub-nanomolar range at the SERT, whereas the binding affinity at the NET seems to be underestimated by one order of magnitude. CoMSIA analysis appears to be performing better when looking at the Ki-values of ADAM (s9) and its fluorine (s20), bromine (s18) and chlorine (s19) analogs. The Ki-values for the NET are predicted in the correct order of magnitude by CoMSIA. However, binding affinity at the SERT seems to be underestimated by one order of magnitude by both CoMFA and CoMSIA. Compound s30, possessing a fluoromethyl group in position 2´ and a iodine atom in position 4´, and being of interest as fluorine-18 labeling of the fluoromethyl group proved to be possible (42), is predicted as displaying SERT binding affinity in the nanomolar range by CoMFA (Ki = 1.13 nM), and in the subnanomolar range by CoMSIA (Ki = 0.69 nM). Binding affinity seems to be slightly underestimated by both methods. Binding affinity at the SERT of ODAM (s16), differing from the other compounds by the oxygen that bridges the two phenyl rings, is also predicted in the correct order of magnitude by both CoMFA and CoMSIA. However, SERT selectivity seems to be slightly overestimated by the models. Binding affinity at the SERT of the pyridinyl compound s17 for which Oya et al. (47) reported only weak binding affinity at the SERT, seems to be wrongly predicted by several orders of magnitude. This, clearly, is a shortcoming of the models. However, most of the tested diphenyl sulfides are correctly predicted by both CoMFA and CoMSIA, thus making the models suitable for the estimation of binding affinities for novel diphenyl sulfide structures.
Bioisosterism
The principle of bioisosteric replacement of functional groups is considered a successful optimization strategy. Langmuir introduced the concept of isosterism in 1919 (48). Isosterism was at that time used to describe the similarity of molecules or ions that have the same number of atoms and valence electrons. This concept was extended by Grimm´s hydride displacement law (49). Grimm termed groups such as OH, NH2 and CH3 pseudoatoms. Friedman (50) introduced the term bioisosterism. Bioisosteres are groups or molecules that are structurally similar and show the same type of biological activity. Comprehensive overviews of isosterism and bioisosterism in drug design are given in literature (51-55). The concept of bioisosterism was used when designing new diphenyl sulfide derivatives.
Design of new compounds
Tables 3 to 5 summarize the molecular structures of the newly designed compounds and their predicted binding affinities at the SERT and at the NET. Very high binding affinity of a radioligand in combination with a comparatively slow clearance from tissue can restrict its usefulness for PET, as the rate-limiting step of tracer retention may become the delivery instead of the binding process (1). A consequence of this would be to only consider substances whose Ki-values are found within a particular range, for instance, between 0.5 and 10 nM as suggested by Lassen et al. (56) for benzodiazepine receptor tracers. However, as the used CoMFA and CoMSIA models cannot predict PET kinetics and thus, no optimum Ki-value for any one compound can be estimated, we did not focus on Ki-values between 0.5 and 10 nM only, but considered substances being estimated as displaying high binding affinity in a nanomolar or subnanomolar range at the SERT and low binding affinity at the NET. Another aspect during molecular design was to obtain structures that could easily be radiolabeled with either carbon-11 or preferably fluorine-18. Carbon-11 labeling of one of the N-methyl groups as seen in [11C]DASB (s12) is not necessarily suitable as the N-methyl groups are susceptible to metabolism. Therefore it seems difficult to quantify radiolabeled metabolites that cross the blood brain barrier and need to be considered during evaluation of a PET study. However, this seems to contrast with recent findings of Halldin et al. (57) who labeled MADAM (s10) with carbon-11 in two different positions (at the methyl group of the phenyl ring, and at one methyl group of the tertiary amino group) and measured metabolism in monkey brain. They found no significant difference with regard to brain kinetics and metabolism, indicating that no radioactive metabolites entered the brain.
When modifying the DASB scaffold, we concentrated on four structural features. These were the substituents in position 2´ and 4´ of ring B, the aromatic ring B itself, and the sulfur bridge between ring A and ring B as shown in Figure 3. From the CoMFA and CoMSIA models it became clear that the substituent in position 4´ should preferably display electron-withdrawing characteristics, which is particularly required for high SERT selectivity. Obviously, a fluoroethyl group in position 4´ as seen in structure s25 does not meet this requirement which explains its selectivity loss compared to DASB (s12) (26). Therefore, the replacement of the fluoroethyl group with a fluoroethyl-oxycarbonyl group (ester) seems promising as the possibility of fluorine-18 labeling is maintained. The estimated Ki-values of compound n4 are 1.23 nM (CoMFA) and 1.43 nM (CoMSIA) for the SERT, and 2239 nM (CoMFA) and 891 nM (CoMSIA) for the NET. Replacement of the fluoroethyl group with a fluoroethyl-carbonyl group (ketone) seems also most promising since compound n27 was predicted to have Ki-values around 0.88 nM (CoMFA) and 0.90 nM (CoMSIA) for the SERT, and around 1622 nM (CoMFA) and 537 nM (CoMSIA) for the NET.
To further improve binding affinity at the SERT, replacement of ring B with an electron deficient pyridine ring seemed reasonable. Depending on the position of the nitrogen atom in the pyridine ring, binding affinity at the SERT is estimated as slightly decreased for compound n64, and as about the same for compound n58, whereas a markedly improved binding affinity at the SERT (Ki = 0.54 nM) is estimated for compound n52 by the CoMFA analysis. Moreover, SERT selectivity seems to be retained, as for compound n52 a Ki-value of 2630 nM at the NET is estimated by using CoMFA. Substitution with a fluoroethyl group in position 4´ as in compounds n50, n56, and n62 appears to result in decreased selectivity, whereas binding affinity at the SERT is predicted in a nanomolar range suitable for a PET ligand. Substitution in position 4´ by a fluoroethyl-carbonyl group as in compounds n53, n59 and n65 results in clearly selective substances. This is demonstrated in Figure 5, showing compound n53 within the SERT CoMFA fields and within the CoMSIA fields retrieved from selectivity analysis. The latter show those areas which are important for selectivity at the SERT over the NET (27). The electron deficient pyridine ring is located in the blue region, which shows that partial positive charges are favorable for high binding affinity at the SERT and for SERT selectivity at this site. Moreover, the fluoroethyl-carbonyl substituent is directed towards the green region. This region denotes areas in which steric bulk enhances binding affinity and SERT selectivity. The oxygen atom of the carbonyl group is directed towards the red region visible in both the SERT CoMFA plot and the CoMSIA selectivity plot. Thus, partial negative charge is located in the red region as required. This may also confirm the choice of the conformation of the fluoroethyl-carbonyl substituent.

Figure 5: Compound n53 within the steric (a) and electrostatic (b) SERT "StDev*Coeff" CoMFA fields, and within the steric (c) and electrostatic (d) "StDev*Coeff" CoMSIA fields from the selectivity analysis. Green regions denote those areas in which steric bulk is favorable for high binding affinity at the SERT and for high SERT selectivity. Yellow regions denote those areas where steric bulk has a detrimental effect on SERT binding affinity and SERT selectivity. Binding affinity and SERT selectivity is enhanced by partial positive charges in blue regions, and by partial negative charges in red regions.
As thiophene is considered a classical bioisostere of benzene (52, 55), the exchange of ring B for thiophene was attempted. Good results were obtained for compound n67 and compound n70, differing from each other in the position of the sulfur atom in the thiophene ring. Both compounds were predicted as displaying high binding affinity at the SERT and low binding affinity at the NET, thus being highly SERT selective. To make fluorine-18 labeling possible, the cyano group was exchanged for the above-mentioned fluoroethyl-oxycarbonyl group, resulting in compounds n69 and n72 which are predicted as also showing high binding affinity at the SERT and as being SERT selective. The only congeneric compounds found in literature with ring B being exchanged by thiophene had been prepared and tested by Sindelar et al. (22). These two compounds displayed only moderate affinity at the SERT. Probably this can be attributed to a different substitution pattern.
As suggested by Oya et al. (42) for compound s30, the amino group in position 2´ can be replaced with a fluoromethyl group without affinity loss and, moreover, this provides a good possibility for fluorine-18 labeling. Affinity and selectivity seem to be retained for compound n12, possessing an electron withdrawing cyano group in position 4´, or for the pyridine compounds n54 and n60. Another electron withdrawing group is the methylsulfonyl group which Burger (52) suggested to be bioisosteric to the trifluoromethyl group. As Wilson et al. (21) found that a trifluoromethyl group in position 4´ makes compound s22 much too lipophilic to display favorable PET kinetics, substitution of position 4´ with a methylsulfonyl group is probably advantageous for a PET ligand. Both compound n21, possessing an amino group in position 2´, and compound n22, possessing a fluoromethyl group in position 2´, are predicted to be highly selective SERT ligands. Compound n21 could possibly be carbon-11 labeled at the methylsulfonyl group, whereas compound n22 can obviously be fluorine-18 labeled. Other possible electron withdrawing substituents in position 4´, probably resulting in selective SERT ligands, are a methylamino-carbonyl group (n25, n26), a methylsulfoxide group (n23, n24), a methylamino-sulfonyl group (n19, n20), a fluoroethyl-thiocarbonyl group (n28), a fluoroethyl-oxythiocarbonyl group (n29), a fluoroethyl-sulfanylcarbonyl group (n30), and a cyanato group (n8). These groups were suggested because of an Optimize QSAR run. Only moderately selective substances seem to result from a thiocyanato group (n9), a methylsulfanyl group (n5) or a trifluoromethylsulfanyl group (n6) in position 4´. A nitroethenyl group in position 4´ (n14) also does not seem to be favorable, although it is suggested to be bioisosteric to halogens or the trifluoromethyl group (55). The idea of placing an additional electron withdrawing group in position 2´ resulted in the suggestion of compounds n15 - n17, all possessing an ester function in position 2´. These compounds are predicted as being highly selective for the SERT. Considering known SARs, these suggestions seem reasonable, which is confirmed by Emond et al. (25) stating that ester functions are generally tolerated in position 2´. Whereas compound n15 can only be carbon-11 labeled, fluorine-18 labeling seems possible for compounds n16 and n17 at either the fluoroethyl group in position 4´, or the fluoromethyl-oxycarbonyl group in position 2´. Substitution of position 2´ with a fluoromethyl-carbonyl group resulted in compound n18 which is predicted to be a particularly selective high affinity SERT ligand as can be concluded from the estimated Ki-values of 0.46 nM (CoMFA) and 0.68 nM (CoMSIA) for the SERT and 6607 nM (CoMFA) and 4074 nM (CoMSIA) for the NET. Moreover, fluorine-18 labeling seems possible. Another idea was to link position 4´ and 5´ with a methylenedioxy group (n43 and n44) in analogy to the SSRI paroxetine (s4), or with a furano ring (n45 -n48). Compounds n44, n46 and n48, possessing a fluoromethyl group in position 2´, are predicted as being as selective as their respective amino analogs n43, n45 and n47. Substitution of position 2´ with a formyl group (aldehyde) (n31 and n36), and their corresponding oximes (n32 and n37), imines (n33 and n38), and methylimines (n34 and n39), was a result of using the Optimize QSAR module. To provide a possibility for fluorine-18 labeling, position 4´ was substituted with either a fluoroethyl group, which in the case of the aldehyde (n31) and the imine (n33) still seems to result in comparatively SERT selective substances, or with a fluoroethyl-oxycarbonyl group, which in all cases seems to result in highly SERT selective compounds. This series of compounds appear also quite convenient for synthesis planning as the oxime, the imine and the methylimine can easily be prepared from the aldehyde. Substitution of position 2´ with a benzoyl rest (n35 and n40), which was also found because of an Optimize QSAR run, seems arguable in some ways. Although it was found by Choi et al. (43) that sometimes quite large substituents are tolerated in position 2´, as for instance a fluorophenyl-carbonylamino group, such sterically demanding substituents are probably too lipophilic to finally display favorable PET characteristics. Another suggestion derived from an Optimize QSAR run is the substitution of position 4´ with a phenyl rest as seen in compound n41 and n42. On the one hand, Emond et al. (25) concluded that sterically demanding groups in position 4´ have got a detrimental effect on binding affinity at the SERT, but on the other hand, such groups at ring B are considered favorable for high SERT affinity and high SERT selectivity by the CoMFA and CoMSIA models. In compound n42, the phenyl ring is further substituted by a fluoromethyl group which could possibly be fluorine-18 labeled.
Starting from the finding of Zhuang et al. (44) that ODAM (s16) is binding with high affinity at the SERT, a series of additional structures were suggested in which the sulfur bridge was replaced with bioisosteric groups or atoms. Classical bioisosteres of divalent sulfur (-S-) are a methylene group (-CH2-), divalent oxygen (-O-), and amine functions (-NH- or -NR-) (52, 55). Less typical, but also known as bioisosteric to divalent sulfur, are a cyanamide (-NCN-) (52, 54, 55, 58) and an ethene group (-CH=CH-) (52, 54, 55). Binding affinity at the SERT is predicted to move towards the sub-nanomolar range for the DASB analogs n77 and n81 in which the sulfur bridge was substituted by either an amine function or a methylene bridge. Binding affinity at the NET seems to increase only slightly and is still in the same order of magnitude as for DASB (s12). Thus, selective SERT ligands can probably be obtained by replacing the sulfur bridge with a methylene bridge or an amine function. DASB analog n73, in which the sulfur bridge is exchanged for an oxygen bridge, seems to show similar binding characteristics as DASB (s12) itself. This is in close agreement with the suggestion of Burger (52) that it is often the steric rather than the electronic properties of the oxygen or sulfur bridge that are the determinants of pharmacological activity. The replacement of the sulfur atom with a (Z)-configurated carbon-carbon double bond results in compound n85, which still seems to show affinity at the SERT in the same order of magnitude as DASB (s12) itself, but is predicted to be markedly less SERT selective. The cyanamide n93 is believed to be highly SERT selective with predicted Ki-values at the SERT of 1.00 nM (CoMFA) and 1.44 nM (CoMSIA), and at the NET of 7079 nM (CoMFA) and 10 715 nM (CoMSIA). These values strongly qualify this compound for inclusion as a possible PET ligand. Moreover, carbon-11 labeling seems possible at the cyanamide group as has been reported for diphenyl[11C]cyanamide (59). Another particularly interesting structure for possible application as PET tracer is compound n97. The amine function bridging the two phenyl rings has been further substituted with a fluoroethyl group that could possibly be fluorine-18 labeled. Just as its ethyl, methyl and fluoromethyl analogs (n96, n94 and n95), compound n97 is predicted to be highly SERT selective. The benzophenone derivative n89 is also predicted to be a highly SERT selective substance with remarkably low affinity at the NET and so are its fluoroethyl-oxycarbonyl analog n91 and its fluoroethyl-carbonyl analog n92. However, binding affinity at the SERT also seems to be lower than for the respective sulfur bridged analogs. Surely, any modifications at the 2´ and 4´ position suggested earlier can be also transferred to the DASB analogs in which the sulfur bridge is replaced by other bioisosteric atoms or groups. This can be interesting for synthesis planning. Yet we feel that compound synthesis and biological testing to verify our predictions is highly desirable before further virtual structure modification.
Conclusion
After thorough investigation of known SARs of SERT ligands, a series of potential new and selective PET radiotracers for the SERT were designed by molecular modeling. As diphenyl sulfides had turned out to be promising PET ligands for the SERT, various modifications were carried out on this scaffold. Many of the newly designed compounds possessing a fluoroethyl-oxycarbonyl group in position 4´ are predicted to be selective high affinity ligands at the SERT and are of particular interest for possible fluorine-18 labeling. The same holds true for compounds possessing a fluoroethyl-carbonyl group in position 4´. Fluorine-18 labeling seems also possible for substances possessing a fluoroethyl group in position 4´, combined with an electron withdrawing group in position 2´ as it is the case for aldehyde n31 and its corresponding oxime n32, imine n33 and methylimine n34, or for compound n18, possessing a flouromethyl-carbonyl group in position 2´. Compound n93 is predicted as highly SERT selective, and the cyanamide substructure may be an alternative for carbon-11 labeling. Compound n97 provides another new option for fluorine-18 labeling. Future studies necessarily need to include synthesis and biological testing of the compounds to confirm the predictions. Although most compounds are predicted as being SERT selective, we acknowledge that only data on SERT and NET affinity had been available when generating the models, whereas data of DAT affinity had been lacking. It further has to be noted that our models do not provide any information concerning PET kinetics. Screening for suitability of the novel compounds as PET ligands is needed. Following this, the generation of models predicting PET kinetics seems conceivable.
Supplementary Material
The molecular structures in SYBYL´s mol2 format are obtainable from the authors.
Acknowlegements
Financial support from the fortüne research program of the University Hospital of Tübingen (732-0-0) is gratefully acknowledged. Special thanks go to everyone from Tripos GmbH, Munich for much appreciated help.
Abbreviations
5-HT, Serotonin; 3D QSAR, Three-Dimensional Quantitative Structure-Activity Relationships; CoMFA, Comparative Molecular Field Analysis; CoMSIA, Comparative Molecular Similarity Indices Analysis; DAT, Dopamine Transporter; GASP, Genetic Algorithm Similarity Program; NE, Norepinephrine; NET, Norepinephrine Transporter; PET, Positron Emission Tomography; PLS, Partial Least Squares; QSAR, Quantitative Structure-Activity Relationships; SAR, Structure-Activity Relationships; SERT, Serotonin Transporter; SPECT, Single Photon Emission Computerized Tomography; SSRIs, Selective Serotonin Reuptake Inhibitors
References
b-carbomethoxy-3-b-(4-(2-fluoroethyl)-3-iodophenyl)-nortropane (FEINT): Synthesis, characterization and in vitro audiography of a potential radioligand for mapping serotonin transporter sites by both PET and SPECT. J Nucl Med, 40 (5 Suppl):306, 1999.
Halldin, C., Gulyas, B., Langer, O. and Farde, L., Brain radioligands - state of the art and new trends. Q J Nucl Med, 45 (2):139-152, 2001.
Scheffel, U., Dannals, R.F., Suehiro, M., Ricaurte, G.A., Carroll, F.I., Kuhar, M.J. and Wagner, H.N., Jr., Development of PET/SPECT ligands for the serotonin transporter. NIDA Res Monogr, 138:111-130, 1994.
Oh, S.J., Ha, H.-J., Chi, D.Y. and Lee, H.K., Serotonin receptor and transporter ligands - current status. Curr Med Chem, 8:999-1034, 2001.
Laakso, A. and Hietala, J., PET studies of brain monoamine transporters. Curr Pharm Design, 6:1611-1623, 2000.
Hume, S.P., Lammertsma, A.A., Bench, C.J., Pike, V.W., Pascali, C., Cremer, J.E. and Dolan, R.J., Evaluation of (S)-[11C]citalopram as a radioligand for in vivo labeling of 5-hydroxytryptamine uptake sites. Nucl Med Biol, 19:851-855, 1992.
Lasne, M.C., Pike, V.W. and Turton, D.R., The radiosynthesis of [N-methyl-11C]-sertraline. Appl Radiat Isot, 40:147-151, 1989.
Shiue, C.-Y., Shiue, G.G., Cornish, K.G. and O'Rourke, M.F., PET study of the distribution of [11C]fluoxetine in a monkey brain. Nucl Med Biol, 22:613-616, 1995.
Smith, D.F., Jensen, P.N., Gee, A.D., Hansen, S.B., Danielsen, E., Andersen, F., Saiz, P.A. and Gjedde, A., PET neuroimaging with [11C]venlafaxine: serotonin uptake inhibition, biodistribution and binding in living pig brain. Eur Neuropsychopharmacol, 7:195-200, 1997.
Farde, L., Halldin, C., Mueller, L., Suhara, T., Karlsson, P. and Hall, H., PET study of [11C]-beta-CIT binding to monoamine transporters in the monkey and human brain. Synapse, 16:93-103, 1994.
Bergstrom, K.A., Halldin, C., Hall, H., Lundkvist, C., Ginovart, N., Swahn, C.G. and Farde, L., In vitro and in vivo characterisation of nor-beta-CIT: A potential radioligand for visualisation of the serotonin transporter in the brain. Eur J Nucl Med, 24 (6):596-601, 1997.
Chen, P., Kilts, C.D., Galt, J.R., Ely, T.D., Camp, V.M., Malveaux, E.J. and Goodman, M.M., Synthesis, characterization and SPECT imaging of serotonin transporter (SERT) with [123I]ZIENT: A new high affinity and selective SERT imaging agent. J Nucl Med, 41 (5 Suppl):39, 2000.
Goodman, M.M., Chen, P., Kilts, C., Ely, T., Hoffman, J.M., Votaw, J. and Camp, V., Radiolabeled 2-
Szabo, Z., Kao, P.F., Scheffel, U., Suehiro, M., Mathews, W.B., Ravert, H.T., Musachio, J.L., Marenco, S., Kim, S.E., Ricaurte, G.A. and et al., Positron emission tomography imaging of serotonin transporters in the human brain using [11C](+)McN5652. Synapse, 20:37-43, 1995.
Szabo, Z., Scheffel, U., Suehiro, M., Dannals, R.F., Kim, S.E., Ravert, H.T., Ricaurte, G.A. and Wagner, H.N., Jr., Positron emission tomography of 5-HT transporter sites in the baboon brain with [11C]McN5652. J Cereb Blood Flow Metab, 15:798-805, 1995.
McCann, U.D., Eligulashvili, V. and Ricaurte, G.A., (+/-)-3,4-Methylenedioxymethamphetamine ('Ecstasy')-induced serotonin neurotoxicity: Clinical studies. Neuropsychobiology, 42:11-16, 2000.
McCann, U.D., Szabo, Z., Scheffel, U., Dannals, R. and Ricaurte, G.A., Positron emission tomographic evidence of toxic effect of MDMA ("Ecstasy") on brain serotonin neurons in human beings. The Lancet, 352:1433-1437, 1999.
Oya, S., Kung, M.-P., Acton, P.D., Mu, M., Hou, C. and Kung, H.F., A New Single-Photon Emission Computed Tomography Imaging Agent for Serotonin Transporters: [123I]IDAM, 5-Iodo-2-((2-((dimethylamino)-methyl)-phenyl)-thio)-benzyl Alcohol. J Med Chem, 42:333-335, 1999.
Oya, S., Choi, S.R., Hou, C., Mu, M., Kung, M.P., Acton, P.D., Siciliano, M. and Kung, H.F., 2-((2-((dimethylamino)methyl)phenyl)thio)-5-iodophenylamine (ADAM): an improved serotonin transporter ligand. Nucl Med and Biol, 27:249-254, 2000.
Tarkiainen, J., Vercouillie, J., Guilloteau, D., Gulyas, B., Sovago, J., Cselenyi, Z., Emond, P., Chalon, S., Sandell, J., Hiltunen, J., Farde, L. and Halldin, C., Carbon-11 Labelling of MADAM in Two Different Positions: A Highly Selective PET Radioligand for the Serotonin Transporter. J Labelled Cpd Radiopharm, 44 (1 Suppl):S193-S195, 2001.
Wilson, A.A. and Houle, S., 2-(Phenylthio)benzylamines as PET serotonin transporter ligands: Synthesis with 11C and evaluation in vivo. J Nucl Med, 41 (5 Suppl):38-39, 2000.
Wilson, A.A., Ginovart, N., Schmidt, M., Meyer, J.H., Threlkeld, P.G. and Houle, S., Novel radiotracers for imaging the serotonin transporter by positron emission tomography: synthesis, radiosynthesis, and in vitro and ex vivo evaluation of [11C]-labeled 2-(phenylthio)araalkylamines. J Med Chem, 43:3103-3110, 2000.
Sindelar, K., Pomykacek, J., Valchar, M., Dobrovsky, K., Metysova, J. and Polivka, Z., Potential antidepressants and inhibitors of 5-hydroxytryptamine and noradrenaline re-uptake in the brain: N,N-dimethyl(arylthio)thenylamines and N,N-dimethyl-2-(thienylthio)benzylamines. Collect Czech Chem Commun, 56:449-458, 1991.
Ferris, R.M., Brieaddy, L., Mehta, N., Hollingsworth, E., Rigdon, G., Wang, C., Soroko, F., Wastila, W. and Cooper, B., Pharmacological properties of 403U76, a new chemical class of 5-hydroxytryptamine- and noradrenaline-reuptake inhibitor. J Pharm Pharmacol, 47:775-781, 1995.
Houle, S., Ginovart, N., Hussey, D., Meyer, J.H. and Wilson, A.A., Imaging the serotonin transporter with positron emission tomography: Initial human studies with [11C]DAPP and [11C]DASB. Eur J Nucl Med, 27:1719-1722, 2000.
Emond, P., Vercouillie, J., Innis, R., Chalon, S., Mavel, S., Frangin, Y., Halldin, C., Besnard, J.-C. and Guilloteau, D., Substituted Diphenyl Sulfides as Selective Serotonin Transporter Ligands: Synthesis and In Vitro Evaluation. J Med Chem, 45:1253-1258, 2002.
Huang, Y., Bae, S.A., Zhu, Z., Guo, N., Hwang, D.R. and Laruelle, M., Fluorinated Analogues of ADAM as New PET Radioligands for the Serotonin Transporter: Synthesis and Pharmacological Evaluation. J Labelled Cpd Radiopharm, 44 (1 Suppl):18, 2001.
Wellsow, J., Machulla, H.-J. and Kovar, K.-A., 3D QSAR of Serotonin Transporter Ligands: CoMFA and CoMSIA studies. Quant Struct-Act Relat, In print, 2002.
SYBYL molecular modeling software, Version 6.6. Tripos Inc.,1699 South Hanley Rd, Suite 303, St. Louis, MO 63144.
Jarkas, N., McConathy, J., Ely, T., Kilts, C.D., Votaw, J. and Goodman, M.M., Synthesis and Radiolabling of New Derivatives of ADAM, Potential Candidates as SERT Imaging Agents for PET. J Labelled Cpd Radiopharm, 44 (1 Suppl):S204-S206, 2001.
Clark, M., Cramer, R.D., III and Van Opdenbosch, N., Validation of the general purpose Tripos 5.2 force field. J Comput Chem, 10:982-1012, 1989.
Jones, G., Willett, P. and Glen, R.C., A genetic algorithm for flexible molecular overlay and pharmacophore elucidation. J Comput-Aided Mol Des, 9:532-549, 1995.
Geladi, P. and Kowalski, B.R., Partial least-squares regression: A tutorial. Anal Chim Acta, 185:1-17, 1986.
Wold, S., Johansson, E. and Cocchi, M., PSL - Partial Least-Squares Projections to Latent Structures, in: Kubinyi, H (eds), 3D QSAR in Drug Design, Theory, Methods and Applications. Vol. Volume 1, ESCOM, Leiden, pp 523-550, 1993.
Cramer, R.D., Patterson, D.E. and Bunce, J.D., Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc, 110:5959-5967, 1988.
Klebe, G., Comparative molecular similarity indices analysis: CoMSIA, in: Kubinyi, H, Folkers, G and Martin, YC (eds), 3D QSAR in Drug Design: Recent Advances. Vol. 3, KLUWER / ESCOM, Dordrecht / Boston / London, pp 87-104, 1998.
Klebe, G., Abraham, U. and Mietzner, T., Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem, 37:4130-4146, 1994.
Owens, M.J., Morgan, W.N., Plott, S.J. and Nemeroff, C.B., Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Ther, 283:1305-1322, 1997.
SYBYL molecular modeling software, Version 6.6, Theory Manual. Tripos Inc.,1699 South Hanley Rd, Suite 303, St. Louis, MO 63144.
Sindelar, K., Pomykacek, J., Holubek, J., Svatek, E., Valchar, M., Dobrovsky, K., Metysova, J. and Polivka, Z., Potential antidepressants and selective inhibitors of 5-hydroxytryptamine re-uptake in the brain: synthesis of several potential metabolites of moxifetin and of two A-ring fluorinated analogs. Collect Czech Chem Commun, 56:459-477, 1991.
Jilek, J., Sindelar, K., Pomykacek, J., Kmonicek, V., Sedivy, Z., Hrubantova, M., Holubek, J., Svatek, E., Ryska, M. and et al., Potential antidepressants: 2-(methoxy- and hydroxyphenylthio)benzylamines as selective inhibitors of 5-hydroxytryptamine re-uptake in the brain. Collect Czech Chem Commun, 54:3294-3338, 1989.
Jilek, J., Urban, J., Taufmann, P., Holubek, J., Dlabac, A., Valchar, M. and Protiva, M., Potential antidepressants. 2-(Phenylthio)aralkylamines. Collect Czech Chem Commun, 54:1995-2008, 1989.
Oya, S., Choi, S.-R., Hou, C., Kung, M.-P., Acton, P.D., Shiue, C.-Y. and Kung, H.F., Synthesis and Characterization of 18F-IDAM as a PET Imaging Agent for Serotonin Transporters. J Labelled Cpd Radiopharm, 44 (1 Suppl):S15-S17, 2001.
Choi, S.-R., Oya, S., Hou, C. and Kung, H.F., Structure-Activity Relationships of analogs of ADAM as Ligands for Serotonin Transporters. J Labelled Cpd Radiopharm, 44 (1 Suppl):S190-S192, 2001.
Zhuang, Z.-P., Choi, S.-R., Hou, C. and Kung, H.F., Synthesis of [123I]ODAM as a Serotonin Transporter Imaging Agent. J Labelled Cpd Radiopharm, 42 (1 Suppl):S357-S359, 1999.
Acton, P.D., Mu, M., Plossl, K., Hou, C., Siciliano, M., Zhuang, Z.-P., Oya, S., Choi, S.-R. and Kung, H.F., Single-photon emission tomography imaging of serotonin transporters in the nonhuman primate brain with [123I]ODAM. Eur J Nucl Med, 26:1359-1362, 1999.
Huang, Y., Bae, S.A., Zhu, Z., Hwang, D.R., Narendran, R., Talbot, P.S., Hacket, E., Kegeles, L.S. and Laruelle, M., Radiosynthesis and evaluation of [18F]AFM, a selective PET tracer for the serotonin transporter. J Nucl Med, 43 (5 Suppl):358P, 2002.
Oya, S., Kung, M.-P., Hou, C., Acton, P.D., Mu, M. and Kung, H.F., Structure-Activity Relationships of IDAM and its Derivatives as Serotonin Transporter Imaging Agents. J Labelled Cpd Radiopharm, 42 (1 Suppl):S57-S59, 1999.
Langmuir, I., Isomorphism, isosterism and covalence. J Am Chem Soc, 41:1543-1559, 1919.
Grimm, H.G., Structure and size of the non-metallic hydrides. Z Elektrochem, 31:474-480, 1925.
Friedman, H.L., Influence of Isosteric Replacement upon Biological Activity, National Academy of Sciences - National Research Council Publication No 206. Vol., Washington D.C., pp 295, 1951.
(Burger, A., A reappraisal of bioisosterism. Pharm Acta Helv, 38:705-709, 1963.
Burger, A., Isosterism and bioisosterism in drug design. Prog Drug Res, 37:287-371, 1991.
Hansch, C., Bioisosterism. Intra-Science Chem Rept, 8 (4):17-25, 1974.
Lipinski, C.A., Bioisosterism in drug design. Annu Rep Med Chem, 21:283-291, 1986.
Thornber, C.W., Isosterism and molecular modification in drug design. Chem Soc Rev, 8:563-580, 1979.
Lassen, N.A., Neuroreceptor quantitation in vivo by the steady-state principle using constant infusion or bolus injection of radioactive tracers. J Cereb Blood Flow Metab, 12 (5):709-716, 1992.
Halldin, C., Tarkiainen, J., Sovago, J., Vercouillie, J., Gulyas, B., Guilloteau, D., Emond, P., Chalon, S., Hiltunen, J. and Farde, L., A PET-comparison in monkey brain of 11C-MADAM labeled in two different positions - selective for the serotonin transporter. J Nucl Med, 43 (5 Suppl):164P, 2002.
Chiu, W.-H., Klein, T.H. and Wolff, M.E., Apparent bioisosteric replacement of -S- by NCN: synthesis of N-cyano-2-aza-A-nor-5.alpha.-androstan-17.beta.-ol acetate, an azasteroid androgen. J Med Chem, 22:119-120, 1979.
Westerberg, G. and Laangstroem, B., Synthesis of [11C]- and [13C]cyanogen bromide, useful electrophilic labeling precursors. Acta Chem Scand, 47:974-978, 1993.
Corresponding Author: Karl-Artur Kovar, Eberhard-Karls-University of Tübingen, Pharmazeutisches Institut, Auf der Morgenstelle 8, 72076 Tübingen, Germany. karl-artur.kovar@uni-tuebingen.de
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps