J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 5(2):181-184, 2002
Single dose pharmacokinetics and bioavailability of glucosamine in the rat
Ali Aghazadeh-Habashi, Saeed Sattari, Franco Pasutto and Fakhreddin Jamali1
Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, CanadaReceived 1 November 2001, Revised 2 May 2002, Accepted 5 June 2002
PDF version
Abstract
Purpose: To study pharmacokinetics of glucosamine following various routes of administration of the hydrochloride salt to rats and locate the site of its first-pass metabolism.
Methods: Rat were cannulated in the jugular vein and single intravenous, oral and intra peritoneal doses of 350 mg/kg were administered. Serial blood samples were collected and plasma glucosamine concentrations were determined using HPLC.
Results: After intravenous administration, the apparent terminal half-life (1.09 ± 0.98 h), apparent steady state volume of distribution (2.1 ± 1.1 L.kg-1) and total body clearance (2.61 ± 0.81 L.kg-1.h-1) were calculated. The peak plasma concentration, after oral administration, occurred approximately 30 min post-dose and the absolute bioavailability was 0.19. Glucosamine was completely bioavailable after intraperitoneal administration.
Conclusion: Orally administered glucosamine is rapidly absorbed, highly distributed and efficiently cleared. The gut rather than liver is mainly responsible for the first pass metabolism since reduced bioavailability is observed after oral but not intraperitoneal doses.
Introduction
Osteoarthritis affects approximately 12% of the general population and the incidence increases with age (1). Current drug therapies, including acetaminophen and nonsteroidal anti-inflammatory drugs, do not slow or reverse the degenerative process in osteoarthritis. Glucosamine (2-amino-2-deoxy-D-glucose) has recently received a great deal of public attention as a treatment for osteoarthritis, prompting scientists to investigate its clinical usefulness and potential adverse effects (1). It has been proposed that glucosamine stops and possibly reverses the degenerative process in osteoarthritis (2). Pharmacokinetic studies of glucosamine in the rat, dog and man have been reported (3-5). However, these investigations have been conducted with radiolabeled drug so that do not differentiate between glucosamine and its metabolites and/or degradation products. We have applied a recently reported specific and sensitive HPLC-UV assay (6), to delineate the glucosamine pharmacokinetics. To explore the site of first pass-metabolism of glucosamine we followed the plasma time course of the drug after administration of single doses to rats via the intravenous (i.v.), intraperitoneal (i.p.) and oral (p.o.) routes.
Materials and Methods
Materials
D-(+)-glucosamine hydrochloride, D-(+)-galactosamine hydrochloride and 1-naphthyl isothiocyanate (>95%) were purchased from Sigma Chemical Company (St. Louis, MO, USA). Methanol and acetonitrile were purchased from Caledon Laboratory Ltd (Georgetown, ON, Canada) while triethylamine and acetic acid were purchased from BDH Inc. (Toronto, ON, Canada). All chemicals and solvents were ACS analytical or HPLC grade. The styrene divinylbenzene quaternary ammonium solid-phase extraction columns (200mg/4.0 mL; particle size range 45 - 150m) were obtained from Alltech Associates, Inc (Deerfield, IL, USA).
Animals
The study protocol was approved by the Health Sciences Animal Policy and Welfare Committee of the University of Alberta.
A group of five male Sprague-Dawley rats (260 ± 4g) was used in i.v. and oral crossover studies with a two-day washout period between the two dosing. The same cannula was used for bolus injection and for withdrawal of blood samples. Following bolus injection, the cannula was washed with normal saline solution to avoid the possibility of carry-over effect. A different group of six rats, in the same weight range, was used in the i.p. study. Rats were housed in cages and maintained in a controlled environment with free access to food and water.
Procedure
Rats were anesthetized with single i.p. injections of sodium pentobarbital (60 mg.kg -1 ), cannulated via the right jugular vein one day prior to the drug administration and fasted overnight. Rats were dosed with exact volumes of glucosamine HCl solution (200 mg.mL -1 in normal saline) to give single doses of 350 mg/kg as i.v. or p.o. injections or as oral gavages. Blood samples (0.3 mL) were drawn at 0, 5, 10, 15, 30, 80, 90, 120, 240, 360, and 480 min post- dose and collected in heparinized tubes. They were centrifuged at 2000 g for 5 min and plasma was harvested and kept at -20° until analyzed.
Analytical Method
The determination of glucosamine in 0.1 mL plasma samples was performed using a previously described HPLC-UV method (6). Galactosamine (internal standard) and glucosamine appeared at 26 and 29 min on chromatogram, respectively. The assay was validated with a detection limit of 0.63 m g.mL -1 and a limit of quantification of 1.25 m g.mL-1. The response was linear over a concentration range of 1.25 - 400 m g.mL -1 and had an intra- and inter-day precision of <11%. Glucosamine was found to be stable in rat plasma at least for one month at -20°. It was also stable at least for 24 h at the room temperature and in automatic injector vials after extraction.
Pharmacokinetic Analysis
Pharmacokinetic indices of glucosamine were determined using the non-compartmental analysis section of WinNolin version 3.1 (Pharsight Corporation, CA, USA). Doses were normalized based on rat body weight. The terminal elimination rate constant, b, for the glucosamine concentration-time curve after i.v. administration was determined by the linear regression of at least three data points from the terminal portion of the plasma concentration-time plots. The area under the plasma concentration-time curves (AUC) was calculated using the trapezoidal rule up to the last measured plasma concentration, Clast . To the latter AUC was added Clast / b to calculate AUC0-· . The total body clearance, ClTB , was determined by i.v. dose divided by AUCi.v . The steady state volume of distribution, Vdss , was calculated from Vdss = dose.MRT/AUCi.v. , where MRT is the mean residence time and AUMC is the area under the moment-time (7). The parameters were determined for each individual animal and their average were calculated. The observed peak plasma concentration (Cmax ) and the time-to-peak concentration (Tmax ) were recorded. Absolute bioavailability (F) after oral doses was calculated as mean of the ratio AUCp.o. /AUCi.v. for every individual rat and for i.p. doses were based on the mean of AUCi.p. divided by mean of AUCi.v. for different groups.
Statistics
All the values are expressed as mean ± standard deviation. Statistical significance between the means of groups was examined using the ANOVA followed by the Duncan Multiple Range test (p<0.05).
Results
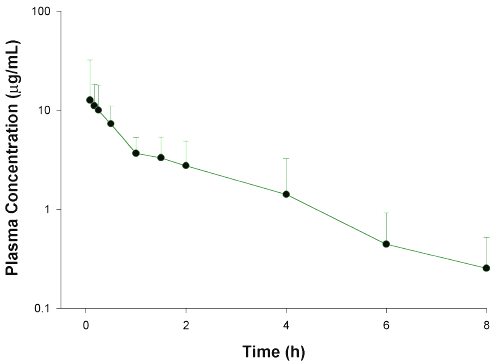
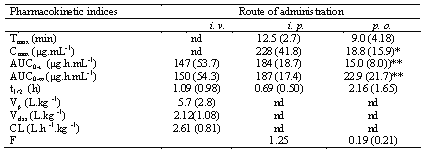
The plasma concentration-time profiles of glucosamine after i.v., i.p., and p.o. doses are shown in Figs.1 and 2, respectively. The mean pharmacokinetic indices are presented in Table 1. After i.v. administration, glucosamine declined in a multiexponential fashion with a rapid initial distribution phase followed by a slower elimination phase (Fig. 1). Absorption after i.p. and oral administrations was rapid as indicated by the occurrence of mea peak plasma concentrations in less than 13 min after both routes of administrations (Table 1). There was no significant difference in the mean AUC values following i.v. and i.p. doses. Following oral doses, however, glucosamine demonstrated a significantly lower mean AUC as compared to both i.v. and i.p. routes. The absolute bioavailability for oral doses was only 0.19± 0.21.

Figure 1: Mean glucosamine plasma concentrations versus time curve after a single i.v. bolus (▲) and i.p. (●) administration of 350mg kg -1 glucosamine HCl to rats. Concentrations below 1.23 mg/mL were not used in the calculation of pharmacokinetic parameters. Error bars represent standard deviation of the mean (n=5 for i.v. and 6 for i.p. ).
Figure 2: Mean glucosamine plasma concentrations versus time curve after a single oral administration of 350mg.kg- 1 glucosamine HCl to rats. Concentrations below 1.23 mg/mL were not used in the calculation of pharmacokinetic parameters. Error bars represent standard deviation of the mean (n=5).
Table 1: Pharmacokinetic indices of glucosamine after administration of 350 mg/kg glucosamine HCl to the rat.
The values are the mean (SD); n = 5 for i.v. and p.o. and 6 for i.p.; nd, not determined;
*significantly different from i.p.; **significantly different from i.v. and p.o.
Discussion
Although glucosamine is considered an endogenous compound, we have not found detectable concentrations in blank plasma of the rat. Following i.v. administration, glucosamine is rapidly distributed and eliminated from the body. The multi-phasic appearance of the log-concentration-time curve indicates a multi-compartment model for the disposition of glucosamine. The observed mean steady state volume of distribution of 2.12 L.kg-1 is rather large and indicates extensive distribution. Glucosamine is rapidly eliminated so that for most samples, the plasma concentration falls below the detectable level of 0.63 g.mL-1 in 6 h, post-dose. In addition to metabolic pathways, previous studies have suggested that glucosamine is taken up by the bone and is incorporated into plasma proteins following administration to the rat and dog (3).
A previous report (3) suggests a much longer terminal t1/2 for glucosamine in the rat. However, since in that study radioabeled drugs was administered differentiation between the unchanged drug and its metabolites was impossible. Nevertheless, the true terminal t1/2 of glucosamine might have been longer that what we have reported herein if the plasma concentration of the drug had been followed for a longer period of time. Under our experimental conditions, however, we were not able to follow drug concentration longer than 6 h due to the limitation in our assay sensitivity. The objectives of our study, i.e., determination of glucosamine bioavailability and the possible site of firs-pass metabolism, however, were achieved.
After oral administration, glucosamine is rapidly absorbed so that some samples taken five and ten minutes after administration contained the highest observed concentrations as compared with the remaining samples (Fig. 2). The oral doses, however, were only 21% bioavailable. This may be attributed to a low gastrointestinal absorption and/or extensive first-pass metabolism. For glucosamine, incomplete oral absorption may be ruled out since following oral administration of radiolabeled glucosamine to the rat, at least 82% of the administered radioactivity has been found to be systemically available (3). Hence, glucosamine is absorbed after oral administration as either unchanged glucosamine or breakdown products. Furthermore, hepatic first-pass metabolism may not be a major component of the overall clearance of glucosamine since i.p. doses exhibit complete bioavailability. Hepatic fist-pass metabolism is expected to affect both p.o. and i.p. doses. The most likely explanation for glucosamine poor bioavailability, therefore, is a substantial loss in the gastrointestinal tract.
Our observations have been made following single doses of 350 mg.kg -1 glucosamine hydrochloride. This dose may be considered rather high as compared with other drugs. However, the recommended dose of glucosamine for the treatment of osteoarthritis is 3 g/day (2). In addition, for pharmacokinetic studies, single oral doses of 6 g have been administered to human volunteers (4).
Conclusion
Glucosamine is rapidly absorbed, highly distributed and efficiently cleared. Since the low bioavailability of the drug is evident only after oral administration, the gut rather than liver is implicated for an apparent large first-pass effect.
Acknowledgement
This study was supported by a National Sciences and Engineering Research Council of Canada # 246039-01 and the Canadian Arthritis Network (National Networks of Excellence).
References
- Hawker G., Update on the epidemiology of the rheumatic diseases. Current Opin Rheumatol. 9: 90-94, 1997.
- Barclay T. S., Tsourounis C., and McCart G. M., Glucosamine, Ann Pharmacother, 32:574-579, 1998.
- Setniker I., Giachetti C., and Zanolo G., Absorption, distribution and excretion of radioactivity after a single dose intravenous or oral administration of [14C] glucosamine to the rat. Pharmatherapeutica, 3: 538-50, 1984.
- Setniker I., Giachetti C., and Zanolo G., Pharmacokinetics of glucosamine in the dog and man. Arznim-forsch., 36: 729-35, 1986.
- Setnikar I., Palumbo R., Canali S., and Zanolo G., Pharmacokinetics of glucosamine in man. Arznim-forsch., 43: 1109-13, 1993.
- Aghazadeh-Habashi A., Sattari S., Pasutto F., and Jamali F., High performance liquid chromatography determination of glucosamine in rat plasma. J Pharm Pharm Sci. Submitted.
- Gibaldi M. and Perrier D., Pharmacokinetics, 2nd edition, Marcel Dekker, Inc. New York, USA, 1982.
Corresponding Author: F.Jamali, Faculty of Pharmacy & Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, Canada, T6G 2N8. fjamali@pharmacy.ualberta.ca
JPPS Contents
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps