J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 5(2):176-180, 2002
High performance liquid chromatographic determination of glucosamine in rat plasma
Ali Aghazadeh-Habashi, Saeed Sattari, Franco Pasutto and Fakhreddin Jamali1
Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, Canada
Received 22 October 2001, Revised 19 April 2002, Accepted 5 June 2002
PDF version
Abstract
Purpose: A high performance liquid chromatographic method was developed for the determination of glucosamine (GlcN) in rat plasma.
Method: Internal standard, galactosamine, was added to 100 mL of plasma containing GlcN followed by precipitation of plasma proteins with acetonitrile. Evaporation of the decanted supernatant solution was accelerated by the addition of methanol. GlcN was derivatized by addition of a solution containing 1-naphthyl isothiocyanate. Sample cleanup included passage through an anion exchange cartridge. Analysis was accomplished by injection of 0.1 mL of the sample solution into an isocratic HPLC system consisting of a C18 column, a mobile phase of acetonitrile: water: acetic acid: triethylamine (4.5: 95.5:0.1:0.05), a flow rate of 0.9 mL/min, and a UV detector set at 254 nm.
Results: Galactosamine and GlcN appeared 26 and 29 min post-injection, respectively. The assay was linear over the range of 1.25-400 mg/mL (CV<10%) with a detection limit of 0.63 μ g/mL and a limit of quantification of 1.25 mg/mL. The method was applied to the determination of GlcN in rat plasma after oral administration of 350 mg/kg of GlcN hydrochloride.
Conclusion: The present assay is specific, sensitive, precise, and accurate and is suitable for pharmacokinetic studies.
Introduction
Osteoarthritis is a degenerative disease of the joint cartilage resulting in a narrowing of the joint space and changes in the underlying bone. It affects approximately 12% of the population in the United States, and the incidence increases with advancing age (1). The incidence is also higher in women than men, with an approximate ratio of two to one. Glucosamine (GlcN), an amino monosaccharide, is a natural component of glycoprotein found in connective tissue and gastrointestinal mucosal membranes. It has recently received a great deal of public attention, prompting scientists to investigate its clinical usefulness and possible adverse effects (1).
Literature methods have been reported in which GlcN was analyzed in nutritional supplements (2), soils (3), and plants (4). Assays for GlcN along with amino acids in bacterial cell walls, glycoconjugate, soya and chitin, after pre-column derivatization, have also been described (5-7). In animal- and human-derived biological samples radiolabeled GlcN has been used for quantitative determination of GlcN and metabolites in pharmacokinetic studies as well as for determining GlcN content in formulations (8, 9). However, these investigations have been conducted with radiolabeled drug and do not differentiate the parent drug from its metabolites and/or degradation products. A pre-column derivatization HPLC method has also been reported for the assay of GlcN in raw material, nutraceutical preparations and dog plasma (10). We have been unable to reproduce this assay due to formation of turbidity and particulate matter in the sample preparation and the presence of interfering peaks in the HPLC chromatograms.
The present work describes a sensitive, precise and accurate HPLC method that uses pre-column derivatization and ion exchange purification for the quantification of GlcN in rat plasma. This method was successfully applied in a pharmacokinetic study in the rat.
Experimental
Materials and reagents
D (+) GlcN hydrochloride, D (+) galactosamine hydrochloride (internal standard) and 1-naphthyl isothiocyanate (>95%) were purchased from Sigma Chemical Company (St. Louis, MO, USA). Methanol and acetonitrile were obtained from Caledon Laboratories Ltd. (Georgetown, ON, Canada) while triethylamine and acetic acid were purchased from BDH Inc. (Toronto, ON, Canada). All chemicals and solvents were of analytical or HPLC grade. The styrene divinylbenzene quaternary ammonium ion exchange cartridges (200 mg/4.0 mL; particle size 45 - 150m) used in sample preparation were obtained from Alltech Associates Inc. (Deerfield, IL, USA). All standard solutions and mobile phases were prepared using double distilled water.
Solutions and standards
A stock solution of GlcN was prepared by dissolving 21.0 mg of GlcN hydrochloride in 25 mL of water (800mg/mL based on free base). Appropriate amounts of the stock solution were diluted with water to give solutions in the range 1.25 - 400mg/mL. These were subsequently used to spike blank plasma for the preparation of plasma standards. The aqueous solutions and plasma standards were freshly prepared prior to use.
Sample preparation and derivatization
Galactosamine (50mL of a 20mg/mL solution) was added to 0.1mL portions of blank rat plasma, which had been spiked with GlcN standard solutions to obtain concentrations of 1.25 to 400mg/mL. Plasma proteins were precipitated by adding 0.4 mL of acetonitrile followed by vortex mixing (one min) and centrifugation (three min) at 10 000 g. The supernatant solution was decanted into a clean test tube, mixed with 0.2 mL of methanol, and evaporated to dryness under vacuum. To the dried residue, derivatizing reagent (0.2 mL of 88 mg/mL 1-naphthyl isothiocyanate dissolved in methanol-acetonitrile-triethylamine, 1:1:0.3) was added and the reaction kept at room temperature for 20 min. The reaction was quenched by adding 0.4 mL of 1.5% acetic acid solution. The excess derivatizing reagent and its degradation products were partitioned into an organic phase by addition of 1.0 mL of chloroform, vortexing for one min, and centrifuging at 2500 g for one min. The upper aqueous layer was transferred to an anion exchange cartridge that had been pre-conditioned by portion-wise washing with 10-15 mL of water. The cartridge was then further washed with 0.5 mL of methanol: water: 1.5% acetic acid (2:1:1) and 0.1 mL of the eluted solution was injected onto the HPLC analytical column.
The recovery of GlcN from rat plasma was established as follows. Three concentrations of spiked plasma samples were analyzed as described above, each in triplicate. A second series of samples was analyzed simultaneously using the same concentration of GlcN in water. Recoveries were calculated by comparing the peak responses for spiked plasma samples with those for samples of GlcN in water. The percent recovery was determined as follows:
% Recovery = Peak response ratio (plasma standard) x 100 / Peak response ratio (water standard)
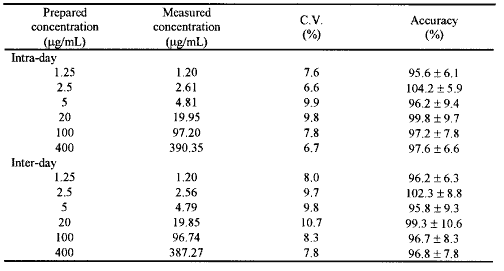
The precision of the method was evaluated by analyzing triplicate spiked plasma samples at concentrations of 1.25, 2.5, 5, 20, 100 and 400mg/mL. Replicate plasma samples (n=3) of six different concentrations were analyzed to determine intra-day precision of the assay. Replicate plasma samples (n=3) were analyzed on three different days to determine inter-day precision of the assay. The accuracy was expressed as the mean % [(mean measured concentration)/(expected concentration)] ∞ 100. The results are shown in Table 1.
Table 1: Intra and inter-day precision (coefficient of variation, CV) and accuracy for glucosamine in rat plasma (n=3).
The stability of GlcN in rat plasma was investigated by adding drug to blank plasma samples to give concentrations of 2.5, 20 and 100mg/mL. These were stored at -20° and aliquots taken at 1 and 6 weeks for analysis.
Chromatographic system
The HPLC system consisted of a model M 45 HPLC pump and model 712 WISP auto-sampler (Waters, Mississauga, ON, Canada), a variable wavelength UV detector model SPD-10A UV-VIS (Shimadzu, Japan), set at 254 nm and 0.02 AUFS for detection, and a model 3390A integrator (Hewlett Packard, USA) for integration.
Analysis was conducted on a 10 cm × 4.6 mm i.d. 5-mm C18 analytical reverse-phase column (Chromatography Sciences Co., St-Laurent, QC, Canada) with a C18 pre-column insert (Waters, Mississauga, ON, Canada). The mobile phase consisted of acetonitrile-water-acetic acid-triethylamine (4.5:95.5:0.1:0.05; pH 4.5), pumped at a flow rate of 0.9 mL/min. A column oven model 1122 / WTC-120 (Waters, Mississauga, ON, Canada) was used to maintain the temperature at 41°.
Animal Study
The study was approved by the Health Sciences Animal Policy and Welfare Committee of the University of Alberta.
One day prior to administration of GlcN, three adult male Sprague-Dawley rats (280-300 g) were anesthetized by i.p. injection of 65 mg/kg sodium pentobarbital. Silastic catheters (0.58 mm i.d. x 0.965 mm o.d.; Clay Adams, Parsippany, NJ) were then surgically implanted into the right jugular vein. The animals were allowed to recover overnight, with free access to water, but were fasted for 12 h prior to drug administration. Single oral doses of 350 mg/kg GlcN hydrochloride were then administered by gavage. Blood samples (0.25 ml) were collected from the jugular vein cannula, into heparinized tubes, at 0, 5, 10, 15, 30 min, 1, 1.5, 2, 4 and 6 h post-dose. Plasma was separated by centrifugation at 10 000 g for 3 min and stored at -20° until analyzed.
Results and Discussion
GlcN, a polyhydroxylated primary amine with polar properties, is difficult to extract from plasma and does not have adequate UV chromophore properties. Extraction and detectability can be improved by appropriate derivatization of the amino functional group. Derivatization using ethyl chloroformate/naproxen, as previously applied in the assay of 2-aryl propionic acids (11, 12), did not give satisfactory results. Pre-column derivatization of amines with 9-fluorenylmethyl chloroformate has been used for the HPLC determination of polyamines (14). Application of this method, using alkaline conditions (borate buffer 0.02 M, pH 9.6), permitted quantitative determination of GlcN in standard aqueous solutions. However, the method was not suitable for rat plasma due to interference from similarly derivatized endogenous amines. Fluorenylmethyl chloroformate derivatives of amino acids, under alkaline or neutral conditions, are anionic due to the presence of an underivatized carboxylic acid group. Under these conditions, the amino acids should remain in the aqueous layer, permitting organic solvent extraction of the derivatized GlcN. While the chromatograms were considerably cleaner, interfering peaks were still evident. Several extraction solvent mixtures (e.g. various composition ratios of hexane/ethyl acetate or chloroform/ethyl acetate) were evaluated for their ability to extract the GlcN derivative with no significant improvements.
Reaction of GlcN with naphthyl isothiocyanate, using a standard aqueous solution, produced a derivative that exhibited favorable UV absorbing properties. This method was subsequently applied to the determination of GlcN in dosage forms (13). In plasma, however, endogenous amino acids and amines also react with naphthyl isothiocyanate resulting in significant background interference in the chromatograms. The addition of excess K 2 CO 3 to `salt out' the interfering endogenous components was successful but also resulted in the removal of GlcN.
Ion-exchange cartridges were introduced into the analytical method in order to take advantage of the anionic character of derivatized endogenous amino acids. Plasma samples were prepared and derivatized with fluorenylmethyl chloroformate as described above with naphthyl isothiocyanate. While this procedure was satisfactory, derivatization with naphthyl isothiocyanate gave much improved chromatograms that were found suitable for the quantification of GlcN in rat plasma.
The naphthyl isothiocyanate derivatization reaction was sensitive to water and several approaches were undertaken to minimize its presence. Protein precipitation, followed by the addition of methanol to the plasma sample, facilitated the vacuum-assisted removal of water. The solvent and concentration of the derivatizing reagent solution were also optimized. A freshly prepared solution of 700 mg of 1-naphthyl isothiocyanate in 8 mL of a mixture of methanol-acetonitrile-triethylamine (3.5:3.5:1) was found to be suitable. Under these conditions the rate of formation of the GlcN derivative was rapid and the reaction complete in 20 min. Excess derivatizing reagent was hydrolyzed by addition of 1.5% acetic acid. The derivatization reaction is shown in Figure 1.
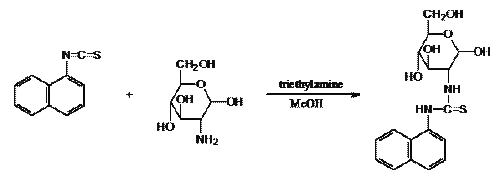
Figure 1: Derivatization of GlcN with 1-naphthyl isothiocyanate.
As depicted in the chromatogram of a spiked plasma sample (Figure 2), the endogenous components of rat plasma eluted before and after the derivatized galactosamine and GlcN peaks appearing at 26 and 29 min respectively. The limit of quantification of GlcN in rat plasma was 1.25mg/mL using 0.1 mL of sample. The intra-day assay (n=3) relative standard deviation at this concentration was 7.6 (Table 1). The detection limit, defined by a signal-to-noise ratio of 3:1, was 0.63 μg/mL. The correlation between drug/internal standard peak area ratio (y) versus concentration of GlcN (X) was linear over the range 1.25 to 400mg/mL (r 2 _ 0.99) with a typical equation of Y = 0.2219 X- 0.015. The accuracy of the assay was >95%, with a CV not exceeding 11%.
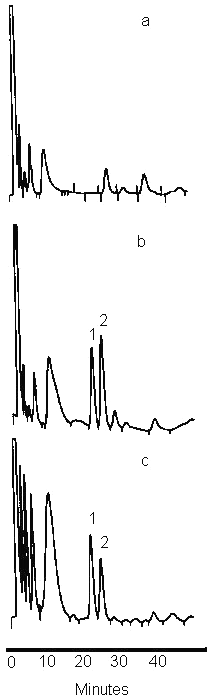
Figure 2: Representative chromatograms of the 1-naphthyl isothiocyanate derivative of GlcN in rat plasma: (a) blank plasma (b), plasma spiked with 20mg/ml of GlcN and (c), plasma sample 90 min after oral dosing of the rat with 350 mg/kg of GlcN hydrochloride. Peaks: 1, internal standard; 2, GlcN.
The stability tests indicated that GlcN is stable in rat plasma for more than one month and the GlcN derivative is sufficiently stable to permit overnight automatic sample injection onto the HPLC system.
The absolute recovery of the assay was 48 ± 3 %. However, a preliminary study indicated that GlcN extraction can be further improved by introduction of the plasma samples into a pre-conditioned anion exchange cartridge, before derivatization, followed by washing the cartridge with methanol-water (1:1). The eluted solution was evaporated to dryness under vacuum and the residue derivatized as previously described.
Daily doses of GlcN as high as 3 g/day are recommended for the treatment of osteoarthritis in humans (15) and pharmacokinetic studies have used single oral doses as high as 6 g (5-6). As a consequence, we studied plasma GlcN concentrations in rat plasma following oral administration of 350 mg/kg of GlcN hydrochloride. The assay was suitable for these studies (Figure 3) but individual samples taken 4 and 6 h post-dose contained GlcN concentrations at or below the assay sensitivity. In these cases larger volumes of plasma were required
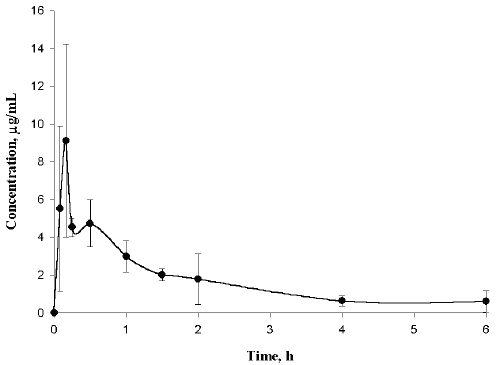
Figure 3: Mean (± SD) plasma concentration versus time profile of GlcN in rat plasma following oral administration of 350 mg/kg of GlcN hydrochloride (n=3).
Conclusion
A specific HPLC method for the determination of GlcN in rat plasma was developed using pre-column derivatization with naphthyl isothiocyanate, and UV detection. The limit of quantification was 1.25μg/mL, which was sufficient for pharmacokinetic studies following administration of oral doses of 350 mg/kg to the rat.
Acknowledgement
This study was supported by the Canadian Arthritis Network (National Centres of Excellence).
References
Hawker G., Update on the epidemiology of the rheumatic diseases. Curr. Opin. Rheumatol., 9: 90-94, 1997.
Wayne K. W., Gibson K. G., and Breite A. G., Determination of glucosamine in nutritional supplements by reversed-phase ion-pairing HPLC. J Liq. Chromatogr. & Rel. Technol., 23: 2861-2871, 2000.
Zhang X. and Amelung W., Gas chromatographic determination of muramic acid, glucosamine, mannosamine, and galactosamine in soils. Soil Biol. Biochem., 28: 1201-1206, 1996.
Osswald W. F., Jehle J., and Firl J., Quantification of fungal infection in plant tissues by determining the glucosamine phenyl isothiocyanate derivative using HPLC techniques. J. Plant Physiol., 145: 393-397, 1995.
Hagen S. R., High performance liquid chromatographic quantitation of phenylthiocarbamyl muramic acid and glucosamine from bacterial cell walls. J. Chromatogr., 632: 63-68, 1993.
Altman F., Determination of amino sugars and amino acids in glycoconjugates using pre-column derivatization with o-phthalaldehyde. Anal. Biochem., 204: 215-219, 1992.
Diaz J., Llibera J. L., Comellas L., and Broto-Puig F., Amino acid and amino sugar determination by derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate followed by high performance liquid chromatography and fluorescence detection. J. Chromatogr. A, 719: 171-179, 1996.
Setnikar I., Giacchetti C., and Zanolo G., Pharmacokinetics of glucosamine in the dog and in man. Arznim-forsch., 36: 729-735, 1986.
Setnikar I., Palumbo R., Canali S., and Zanolo G., Pharmacokinetics of glucosamine in man. Arznim-forsch., 43: 1109-1113, 1993.
Liang Z., Leslie J., Adebowale A., Ashraf M., and Edington A. D., Determination of the nutraceutical, glucosamine hydrochloride, in raw material, dosage forms and plasma using pre-column derivatization with ultraviolet HPLC. J. Pharm. Biomed. Anal., 20: 807-814, 1999.
Wright M. R., Sattari S., Brocks D. R., and Jamali F., Improved high performance liquid chromatographic assay of ibuprofen. J. Chromatogr., 589: 259-265, 1992.
Sattari S. and Jamali F., Involvement of the rat gut epithelial and muscular layer and microflora in chiral inversion and acylglucurondation of R-fenoprofen. Eur. J. Drug Metab. Pharmacokin., 22: 97-110, 1997.
Russell A. S., Aghazadeh-Habashi A., and Jamali F., Active ingredient consistency of commercially available glucosamine sulfate products. J. Rheumatol. In press.
Sabri M. I., Soiefer A. I., Kisly G. E., and Spencer S., Determination of polyamines by pre-column derivatization with 9-fluorenylmethyl chloroformate and reverse phase high performance liquid chromatography. J. Neurosci. Meth., 29: 27-31, 1989.
Barclay T. S., Tsourounus C., and McCart G. M., Glucosamine, Ann. Pharmacother. 32: 574-579, 1998.
Corresponding Author: F.Jamali, Faculty of Pharmacy & Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, Canada, T6G 2N8. fjamali@pharmacy.ualberta.ca
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps