J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 4(3):217-227, 2001
Inhibition of human CYP3A4 activity by grapefruit flavonoids, furanocoumarins and related compounds
Ping-Chuen Ho, Dorothy J. Saville
School of Pharmacy, University of Otago, Dunedin, New ZealandSompon Wanwimolruk1
College of Pharmacy, Western University of Health Sciences, Pomona, California, USAReceived March 30, 2001, Revised August 23, 2001, Accepted August 24, 2001
PDF Version
Abstract
PURPOSE. To evaluate the inhibition of CYP3A4 activity in human liver microsomes by flavonoids, furanocoumarins and related compounds and investigate possibly more important and potential inhibitors of CYP3A4 in grapefruit juice.
METHODS. The effects of various flavonoids and furanocoumarin derivatives on CYP3A4 activity in two human liver microsomal samples was determined using quinine as a substrate. All flavonoids and furanocoumarin derivatives were dissolved in DMSO. In all cases, inhibition activities were compared with activities in control incubations containing 0.2% (v/v) DMSO.
RESULTS. The results showed that the inhibition of quinine 3-hydroxylation (CYP3A4 activity) by bergapten (67%), and quercetin (55%) was greater than naringenin (39%) and naringin (6%), at the same inhibitor concentration of 100 M. The results also demonstrated that the furan ring in the furanocoumarins enhanced the inhibitory effect on CYP3A4 activity. Flavonoids with more phenolic hydroxyl (-OH) groups produced stronger inhibition than those with less hydroxyl groups. Of all the chemicals studied, bergapten (5-methoxypsoralen) with the lowest IC50 value (19-36 mM) was the most potent CYP3A4 inhibitor.
CONCLUSIONS. These results suggest that more than one component present in grapefruit juice may contribute to the inhibitory effect on CYP3A4. Bergapten appears to be a potent inhibitor of CYP3A4, and may therefore be primarily responsible for the effect of grapefruit juice on CYP3A4 activity.
Introduction
Flavonoids are polyphenolic compounds with antioxidant properties and are widely distributed in foods of plant origin such as vegetables, fruit, tea and wine (1). When administered orally, natural flavonoids such as flavone, tangeretin, and nobiletin can activate rat and human benzo(a)pyrene hydroxylase and some other cytochromes P450 (CYP) activities (2). Interaction between grapefruit juice and clinically used drugs has been reported in recent years. These drugs include cyclosporine, midazolam, triazolam, and calcium antagonists such as felodipine, nifedipine, nitrendipine and nisoldipine (3 and references therein). All of these drugs are substrates for CYP3A4 and undergo extensive metabolism by intestinal CYP3A4 (4-6). CYP3A is the largest subfamily of CYP enzymes expressed in the human liver and gastrointestinal tract (7,8) and is involved in the metabolism of many clinically used drugs and other chemicals. Although the inhibitory effects of grapefruit juice on human CYP3A4 are well documented, its effect on other CYP isoforms remains unclear. Previous in vivo studies showed that elimination half-lives of caffeine and coumarin were prolonged by co-ingestion with grapefruit juice. These reactions are considered to be mediated mainly by CYP1A2 and CYP2A6, respectively (9-11).
Interaction between drugs and grapefruit juice results in an increase in the oral bioavailability of drugs. For instance, the consumption of grapefruit juice led to a marked reduction of CYP3A4 activity in the small intestine and was associated with a five times increase in felodipine Cmax and tripled mean area under the curve (AUC) [3 and references therein]. However, the active components responsible for in vivo inhibition of CYP3A4 activity have yet to be fully determined. The predominant flavonoid in grapefruit juice is naringin and is likely to be one of the components in grapefruit that affects drug metabolism. In vitro studies reveal that flavonoids can inhibit microsomal oxidation of felodipine as well as nifedipine (12,13). However, naringin appears to be a weak inhibitor of microsomal felodipine oxidation and its aglycone, naringenin, may be a much more potent inhibitor (12). An in vivo study has demonstrated that the increase in felodipine AUC by naringin solution was much less than that observed with grapefruit juice, indicating that other factors were important (14). Further investigation using a naringin capsule formulation resulted in no change in mean or individual nisoldipine pharmacokinetics compared with water (15). A similar phenomenon occurs with quercetin, which is an inhibitor of CYP3A4. No in vivo inhibition of CYP3A4 mediated metabolism of nifedipine is produced after ingestion of a high dose of quercetin (16).
Recent studies have shown that in addition to flavonoids, other compounds in grapefruit juice might be involved in the drug interaction. A furanocoumarin, 6',7'-dihydroxybergamottin was currently found to be a potent inhibitor of CYP3A4 in liver microsomes (17). Several components of grapefruit juice were isolated from extracts with ethyl acetate and found to be inhibitors of human liver microsomal CYP3A4. These compounds were identified as furanocoumarin derivatives (18). Later, analysis of ethyl acetate extracts from grapefruit juice revealed the presence of several furanocoumarins of which bergamottin, the parent compound of 6',7'-dihydroxybergamottin, is the major one and was found to be a mechanism-based inactivator of CYP3A4 in human liver microsomes (19). Other components of grapefruit juice may also contribute to the interaction with CYP3A4. The other non-flavonoid components found in grapefruit juice such as limonin and obacunone, a triterpene-derived product, also reduced microsomal testosterone 6b-hydroxylation in human liver (18).
Thus the present study was conducted to examine the effect of flavonoids and furanocoumarin compounds found in grapefruit, on the activity of human liver CYP3A4. Even though 6',7'-dihydroxybergamottin is considered to be an important inhibitor, it is not commercially available and was not included in the current study. This investigation also evaluated the effect of coumarin derivatives because we intended to determine whether coumarins have a different effect from the furanocoumarin derivatives. Quinine was selected as a probe for CYP3A4 as it has been demonstrated to be primarily metabolised by human CYP3A4 (20).
Methods and Materials
Chemicals and Reagents
All chemicals and reagents were of analytical grade and distilled water was MilliQ® filtered. Sodium dodecyl sulphate, HPLC-grade acetonitrile and methanol were obtained from BDH Chemicals (Poole, UK). Dimethylsulphoxide (DMSO), tetrabutylammonium bromide, NADPH, bovine serum albumin, flavonoids and coumarin derivatives including, rutin, morin, myricetin, hesperidin, hesperetin, neohesperidin, kaempferol, naringenin, naringin, quercetin, rhoifolin, apigenin, scopoletin, coumarin, 7-ethoxycoumarin, umbelliferone, bergapten, methoxalen and psoralen were purchased from Sigma Chemical Co (St. Louis, MO, USA). Fisetin, galangin, chrysin, and flavone were obtained from Aldrich (Milwaukee, WI, USA). Prunin was purchased from Roth (Karlsrule, Germany). Narirutin was purchased from Indofine (Belle Mead, NJ, USA). Quinine hydrochloride dihydrate was purchased from Merck-Schuchardt (Hohenbrunn, Germany). 3-Hydroxyquinine was a gift from Dr. P Winstanley, University of Liverpool, UK.
Preparation of Human Liver Microsomes
Two human livers used were from Caucasian male donors aged 50 years who had met traumatic deaths. They were neither taking medication nor had significant past medical history. The use of human livers was approved by the Southern Regional Health Authority (Otago) Ethics Committee, Dunedin, New Zealand. Human liver microsomes were prepared by a standard differential ultracentrifugation as previously described (20). The microsomal protein concentration was determined using a BCA reagent as described by the manufacturer (Sigma Chemical Co., St. Louis, MO, USA) and bovine serum albumin was used as a standard.
Inhibition Experiments
The effects of various flavonoids and furanocoumarin derivatives on quinine metabolism were determined in microsomes from two human livers (HL1 and HL2). It has been proposed that grapefruit juice inhibits drug metabolism by selective downregulation of the intestinal CYP3A4 and has no effect on hepatic CYP3A4 (5). The use of liver microsomes instead of intestinal microsomes is still valid as it has been shown that the human CYP3A4 forms expressed in liver and intestine are identical (21). Hence, observations made using hepatic microsomes should be readily applicable to intestinal metabolism. Quinine was used as a substrate at a concentration of 100 mM, approximating the apparent Km value for quinine 3-hydroxylation determined previously in these livers. The potential inhibitors were studied at three concentrations (10, 100, and 200 mM) chosen to be selective for the respective CYP isoforms on the basis of published Ki or Km values (13, 22). All inhibitors were dissolved in DMSO. In all cases, inhibited activities were compared with activities in control incubations (a final volume of 0.25 ml) containing 0.2% (v/v) DMSO as appropriate.
IC50 values (i.e. inhibitor concentration which produces 50% inhibition) were estimated using the non-linear regression modelling programme, Minim® (courtesy of Dr. R Purves, Department of Pharmacology, University of Otago, Dunedin, New Zealand) by fitting the following equation (23), where [I] was a concentration of inhibitor:
Human microsomes (1 mg/ml) were incubated with quinine (100 mM) in the presence and absence of inhibitors with 1 mM NADPH in phosphate buffer (0.067 M, pH 7.4) at a final volume of 0.25 ml. Each incubation was performed in duplicate. The reaction was initiated by the addition of NADPH. Incubations were performed at 37°C in a shaking water bath for 15 min. The reaction was terminated by the addition of cold methanol solution (0.5 mL). The samples were vortexed briefly and centrifuged at 2500 g for 10 min. The resultant supernatant (30 mL) was injected onto the HPLC column.
HPLC Assay for 3-Hydroxyquinine
The major metabolite of quinine, 3-hydroxyquinine (3-OHQ) was assayed by a reversed-phase HPLC method (24). The detection limit of the method was 0.1 M (0.034 mg/ml). The inter- and intra-assay coefficient of variation was less than 7% over the concentration range of 0.1 to 30 mM.
Results
The effects of 10 flavones found in grapefruit, on quinine 3-hydroxylase (i.e. CYP3A4) activity in human liver microsomes were evaluated (Table 1).
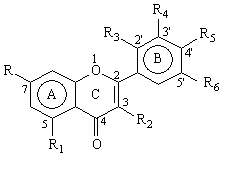
These compounds exhibit similar structures with different hydroxyl (OH) substitutions. The results show that two flavone glycosides, rutin and rhoifolin did not inhibit the metabolism of quinine at low and medium concentrations of the inhibitors (10 and 100 mM). Only a moderate inhibition was observed with a 200 mM concentration of these two glycosides. The other 8 flavones produced considerable inhibition in CYP3A4 activity, which was dependent on their concentration and chemical structures. Myricetin was the most potent inhibitor among these flavones whereas apigenin appeared to be a weak inhibitor. As the number of hydroxyl substitutions increased, the inhibition of CYP3A4 activity tended to be progressively enhanced. At a concentration of 100 mM, flavone (without any hydroxyl group) had little effect, and apigenin (with 3 hydroxyl groups) caused 30-37% inhibition. In contrast, morin and quercetin (with 5 hydroxyl groups) produced 34-41% and 55-65% inhibition respectively, and myricetin (with 6 hydroxyl groups) had 78-94% inhibition (Table 1). The inhibitory potency appeared to be in order from flavone < apigenin < kaempferol < chrysin < morin < fisetin < quercetin < myricetin. The hydroxyl groups at R4 position on the B ring could enhance the inhibitory potency of flavonoids. For example, both fisetin and kaempferol have four hydroxyl groups but the extent of CYP3A4 inhibition was different. At the concentration of 100 mM, fisetin (fourth hydroxyl group at R4 on the B ring) caused 71-73% inhibition whereas kaempferol (fourth hydroxyl group at R1 on the A ring) caused only 30-41% inhibition (Table 1). A similar phenomenon was observed with quercetin (fifth hydroxyl group at R4 on the B ring) which caused 55-65% inhibition at concentration of 100 mM while morin, with the fifth hydroxyl group at R3 position on the B ring, produced only 34-41% inhibition.
Table 1: Structures of flavones investigated and their inhibition of quinine 3-hydroxylation (CYP3A4 activity) in two human liver microsomes, HL1 and HL2. The inhibitors were studied at three concentrations, 10, 100 and 200 mM. Each value represents the average of two separate determinations that differed by < 10%.
% Inhibition of quinine
3-hydroxylation
R
R1
R2
R3
R4
R5
R6
10mM
100mM
200mM
Flavone
H
H
H
H
H
H
H
HL1
-14.1
13.3
40.6
HL2
-6.2
22.3
45
Chrysin
OH
OH
H
H
H
H
H
HL1
19.5
63.3
51.6
HL2
43.1
56.4
56.4
Apigenin
OH
OH
H
H
H
OH
H
HL1
10.9
29.7
27.1
HL2
18.5
37.4
38
Fisetin
OH
H
OH
H
OH
OH
H
HL1
14.1
71.1
70.3
HL2
18.5
72.5
74.4
Kaempferol
OH
OH
OH
H
H
OH
H
HL1
2.3
41.4
41.4
HL2
18.5
29.9
43.1
Morin
OH
OH
OH
OH
H
OH
H
HL1
4.7
34.4
51.6
HL2
15.6
41.2
60.2
Quercetin
OH
OH
OH
H
OH
OH
H
HL1
21.1
54.7
61.7
HL2
28
64.9
63
Myricetin
OH
OH
OH
H
OH
OH
OH
HL1
64.1
78.1
75.8
HL2
82.5
93.6
90.3
Rutin
OH
OH
-O-rutinose
H
OH
OH
H
HL1
0.8
-10.9
-5.5
HL2
-1.4
9
26.1
Rhoifolin
R-G-a
OH
H
H
H
OH
H
HL1
-0.8
6.3
18.0
HL2
1.4
10.9
26.1
a rhamnose-glucose-, L-rhamnose is linked a 1 ® 2 to D-glucose
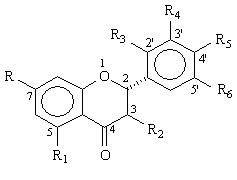
The effects of the flavanone compounds found in grapefruit, on the 3-hydroxylation of quinine (CYP3A4 activity) are summarised in Table 2.
Table 2: Structures of flavanones used and their inhibition of quinine 3-hydroxylation (CYP3A4 activity) in two human liver microsomes, HL1 and HL2. The inhibitors were studied at three concentrations, 10, 100 and 200 mM. Each value represents the average of two separate determinations that differed by < 10%.
% Inhibition of quinine
3-hydroxylation
R
R1
R2
R3
R4
R5
R6
10mM
100mM
200mM
Galangin
OH
OH
OH
H
H
H
H
HL1
21.1
50
51.6
HL2
28.9
54.5
49.8
Hesperetin
OH
OH
H
H
OH
OCH3
H
HL1
-8.6
28.9
51.6
HL2
9
35.5
58
Naringenin
OH
OH
H
H
H
OH
H
HL1
8.6
39.1
64.1
HL2
-5.7
34.6
46.9
Naringin
R-G-a
OH
H
H
H
OH
H
HL1
-5.5
6.3
14.1
HL2
23.3
48.9
46.9
Neohesperidin
R-G-a
OH
H
H
OH
OCH3
H
HL1
7.8
17.2
38.3
HL2
-2.4
8.5
30.3
Hesperidin
R-G-b
OH
H
H
OH
OCH3
H
HL1
-4.7
6.3
0
HL2
-4.3
20.4
24.2
Narirutin
R-G-b
OH
H
H
H
OH
H
HL1
1.2
-6.3
3.9
HL2
-7.6
-1.4
2.4
Prunin
Glucose-
OH
H
H
H
OH
H
HL1
14.8
20.3
21.9
HL2
18.5
26.1
14.7
b a rhamnose-glucose-, L-rhamnose is linked a 1 ® 2 to D-glucose
rhamnose-glucose-, L-rhamnose is linked a 1 ® 6 to D-glucose
At low concentration (10 mM) of the flavanones, there was no inhibitory effect, except for galangin, naringin, and prunin. When the concentrations of the flavanones were increased to 100 mM and 200 mM, all compounds (except narirutin) inhibited the activity of CYP3A4. Two flavanone glycosides naringin and neohesperidin produced less inhibitory effect than their corresponding flavanones naringenin and hesperetin. At 200 mM concentration, hesperetin, and naringenin caused a similar extent of inhibition ranging from 47 to 64% (Table 2). Neohesperidin and naringin were weak inhibitors of CYP3A4 as only 14 to 38% inhibition was observed (Table 2). Hydroxyl substitution at R2 position clearly enhanced the inhibitory effect. For example, at low and medium concentrations (10 and 100 mM) galangin, with a hydroxyl group at R2 position, showed greater inhibition than hesperidin and naringenin. Table 3 shows inhibitory effects of four different coumarin derivatives on the activity of CYP3A4.
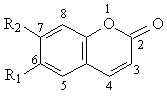
Table 3: Structures of coumarin derivatives and their inhibition of quinine 3-hydroxylation (CYP3A4 activity) in two human liver microsomes, HL1 and HL2. The inhibitors were studied at three concentrations, 10, 100 and 200 mM. Each value represents the average of two separate determinations that differed by < 10%
% Inhibition of quinine
3-hydroxylation
R1
R2
10mM
100mM
200mM
Coumarin
H
H
HL1
-7.0
-15.6
-20.3
HL2
7.1
-15.6
-13.7
7-Ethoxycoumarin
H
CH3CH2O-
HL1
5.5
0.8
3.9
HL2
3.3
-6.2
-3.3
Umbelliferone
H
OH
HL1
7.8
10.9
22.7
HL2
8.1
20.4
41.2
Scopoletin
CH3O-
OH
HL1
-3.1
28.9
35.9
HL2
23.2
52.6
50.7
Coumarin and 7-ethoxycoumarin had no effect on the CYP3A4 activity, at the three concentrations studied whereas, umbelliferone and scopoletin produced moderate inhibition of CYP3A4 activity at their medium and high concentrations (100 mM and 200 mM). It was noted that hydroxyl substitution increased the inhibitory effect, as indicated by comparison of coumarin and umbelliferone (Table 3). Further study with furanocoumarin derivatives has shown that they produced a greater degree of inhibition than those observed with coumarin derivatives (Table 4).
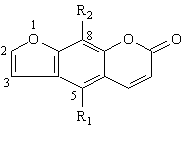
Bergapten, methoxalen and psoralen were found to be moderate to potent inhibitors of CYP3A4. Their inhibitory effects were increased with increasing concentrations of the furanocoumarins.
Based on the data presented in Tables 1 to 4, the IC50 values were estimated for individual compounds and these are summarized in Table 5.
Table 4: Structures of furanocoumarin derivatives used and their inhibition of 3-hydroxylation of quinine (CYP3A4 activity) in two human liver microsomes, HL1 and HL2. The inhibitors were studied at three concentrations, 10, 100 and 200 mM. Each value represents the average of two separate determinations that differed by < 10%.
% Inhibition of quinine
3-hydroxylation
R1
R2
10mM
100mM
200mM
Bergapten
CH3O
H
HL1
33.6
67.2
64.8
HL2
43.1
67.8
71.6
Methoxalen
H
CH3O
HL1
26.6
71.1
78.1
HL2
9.0
82.7
92.9
Psoralen
H
H
HL1
1.6
43.8
71.1
HL2
3.8
9.5
42.2
Among the compounds studied, bergapten and methoxalen were the most potent inhibitors of CYP3A4, with IC50 values of 19-36 mM and 35-39 mM, respectively (Table 5). Flavonoids including quercetin, fisetin, chrysin, naringenin, and galangin were found to be moderate CYP3A4 inhibitors with IC50 ranging from 41 to 136 mM. Naringin and hesperidin were poor inhibitors of CYP3A4 with IC50 values of 1349 and 601 mM, respectively.
Discussion
The present study has demonstrated a dose-dependent inhibitory effect of some flavones, flavonones, coumarin and furanocoumarin derivatives on the activity of CYP3A4 in two human liver microsomes. The extent of inhibition on CYP3A4 produced by grapefruit flavonoids and furanocoumarins, e.g. kaempferol, quercetin, naringenin and bergapten, is similar to that observed with grapefruit juice. At a level of 2.5% grapefruit juice with human liver microsomes produced 40-70% inhibition of CYP3A4 activity (25).
Table 5: Derived IC50 values for the inhibition of 3-hydroxyquinine formation by flavonoids and coumarin derivatives
IC50 (mM)
Inhibitors
Human Liver 1
Human Liver 2
Flavanone (glycosides):
Neohesperidin
280
291
Hesperidin
n.d.
601
Naringin
1349
n.d.
Narirutin
n.d.
n.d.
Prunin
n.d.
n.d.
Flavanone (aglycones):
Galangin
136
117
Hesperetin
175
163
Naringenin
139
188
Falvone (glycosides):
Rhoifolin
n.d.
398
Rutin
n.d.
354
Flavone (aglycones):
Flavone
224
214
Chrysin
86
70
Fisetin
53
44
Kaempferol
223
540
Morin
188
135
Quercetin
82
41
Myricetin
n.d.
n.d.
Apigenin
n.d.
n.d.
Furanocoumarin derivatives:
Bergapten
36
19
Methoxalen
35
39
Psoralen
116
257
Coumarin derivatives:
Scopoletin
307
145
Umbelliferone
543
272
Coumarin
n.d.
n.d.
7-Ethoxycoumarin
n.d.
n.d.
n.d.= not determined, as the data could not be fitted by non-linear regression analysis or these compounds caused no inhibitory effect
There was no inhibition of CYP3A4 observed with glycosides of flavones (rutin and rhoifolin). Rutin and rhoifolin are the rutinose and rhamnose-glucose derivatives of quercetin and apigenin, respectively (Table 1). They are much more water soluble than their corresponding flavones (26). It is possible that the high polarity of rutin and rhoifolin may have interfered with their interaction with the CYP enzyme. This also suggests that the hydroxyl substitution on ring A is an important functional group for the inhibitory effect on CYP3A4. Similar observations were seen with glycosides of flavanones (neohesperidin, hesperidin, narirutin and prunin, Table 2).
Saturation of the C2-C3 double bond in flavanones (Table 2) caused a decrease in inhibitory potency on CYP3A4 activity compared with flavones. Flavones have been described as fairly planar molecules whereas flavanones are bulkier molecules. Thus the conformation of the molecule is probably important (27).
Previous studies have shown that the numbers and positions of hydroxyl groups on the A and B rings of flavonoids are important for the effects on enzyme activities (12,26). It has been proposed that the 7-hydroxyl group preferentially interacts with Fe(III) of CYP (28). Consistent with this, our study has shown that compounds without a free hydroxyl substituent on ring A at the R position had no inhibitory effect, while those with a hydroxyl group were good inhibitors (Table 1). The lower inhibitory effect of naringin, with IC50 value 10-fold greater than that of naringenin, could be due to the absence of a free hydroxyl group at the R position on ring A (29). It is known that the 7-hydroxyl group of flavonoids is the first to dissociate and is thus the most likely site of attack by the peroxy radical (30). Similar differences were observed between hesperidin and hesperetin. The IC50 value of hesperidin was 3.7-fold higher than that of hesperetin (Table 5). Interestingly, this inhibitory effect mimicked that observed with the inhibition of 11b-hydroxysteroid dehydrogenase in guinea pig kidney microsomes (31). The IC50 value of naringin (about 2400 mM) was approximately 8-fold higher than that of naringenin (about 340 mM) (24). Lee et al. (32) also reported that the IC50 value of naringin was much greater than naringenin.
The results also show that the inhibitory effect of flavones is related to their molecular structures. Flavones having more hydroxyl substitutions showed stronger inhibition than those with fewer hydroxyl groups. The order of potency is myricetin > quercetin/morin > kaempferol > apigenin > flavone (with 6, 5, 4, 3, 0 hydroxyl groups respectively, Table 1). This was confirmed by the existence of a positive significant correlation (r = 0.89, p < 0.0005) between number of hydroxyl groups and extent of CYP3A4 inhibition of these six compounds at a concentration of 200 mM. This finding is in agreement with a previous study showing that the ability of flavonoid compounds either to inhibit or stimulate benzo(a)pyrene hydroxylation in human liver microsomes was related to their possession or lack of hydroxyl group respectively (33). The order of inhibitory potency for fisetin (with 4 hydroxyl groups) and chrysin (with 2 hydroxyl groups) were not line in with the mentioned flavones. Fisetin had greater effect than kaempferol which has the same number of hydroxyl groups (Table 1). Chrysin produced more inhibitory effect than apigenin (with 3 hydroxyl groups), even though it has less hydroxyl groups. When the data from fisetin and chrysin were included in the correlation analysis with those six flavone compounds, there was no significant correlation (r = 0.36, p > 0.1) between number of hydroxyl groups and extent of CYP3A4 inhibition. This suggests that number of hydroxyl groups and position of hydroxyl substitutions are both important. Quercetin has also been reported to be a more effective inhibitor of CYP3A4-catalysed oxidation of felodipine and nifedipine than naringenin and kaempferol. Their order of potency is quercetin > kaempferol > naringenin (with 5, 4, 3 hydroxyl groups respectively) (13).
Furanocoumarin derivatives produced a greater inhibition effect than coumarins indicating that the furan ring increased the potency of inhibition of CYP3A4. Bergapten (5-methoxypsoralen), a furanocoumarin, is present in grapefruit peel oil (34) and some plant foods such as parsley, parsnips and celery (35). In our study, bergapten appears to be the most potent inhibitor of CYP3A4 with IC50 value of approximately 25 mM (19 and 36 mM). The IC50 value of bergapten was similar to the IC50 of bergamottin (22 mM) but 10-fold higher than that of 6',7'-dihydroxybergamottin (2 mM) [25]. Bergapten was approximately 6-fold more potent than naringenin and 50-fold more potent than naringin. This suggests that bergapten may be one of the important components in grapefruit juice which is responsible for the interactions between drugs and grapefruit juice. Several other furanocoumarins have previously been reported to cause mechanism-based inactivation of CYP, e.g., corandrin (36) and methoxalen (8-methoxypsoralen) [37, 38]. 6',7'-Dihydroxybergamottin and its parent compound, bergamottin, found in grapefruit juice have also been shown to cause mechanism-based inactivation of CYP3A4 (17,19). Bergapten was also shown to produce mechanism-based inhibition of CYP3A4 (39). All these compounds contain a furan ring, which has been suggested to be the group responsible for the inactivation of CYP1A based on studies of a series of naturally occurring coumarins (40). Several furans are known to undergo metabolic activation to form reactive epoxide intermediates (41,42). It has been reported that the active metabolites of 8-methoxypsoralen bind covalently to microsomal proteins and inactivate CYP (43). This may imply that diets such as grapefruit juice contribute to the alteration of activity of liver and intestinal CYP3A isoforms through both direct inactivation and inhibition (18).
Grapefruit juice contains many flavonoids and furanocoumarin derivatives that may alter the metabolism of drugs by CYP. Of the flavonoids, naringin is the predominant constituent in grapefruit juice, and presents in concentrations up to 1200 mg/L (2000 mM) [3,44]. Naringenin has been detected in grapefruit juice at a concentration of 240 mg/L (880 mM) [44-46]. Grapefruit juice contains a lower concentration of narirutin with up to 120 mg/L (205 mM) [45]. Quercetin has been found in fresh grapefruit juice at a concentration of 5-6 mg/L or 20 mM (22). Very little hesperidin (up to 16 mg/L or 26 mM), neohesperidin (up to 10 mg/L or 16 mM), and hesperetin (up to 3 mg/L or 10 mM) have been detected in grapefruit juice (45). The concentration of the furanocoumarin, 6',7'-dihydroxybergamottin, in grapefruit juice has been found to average 30 mM or 12 mg/L (17, 25). Bergapten has been detected in grapefruit juice at concentrations up to 7 mg/L (30 mM) [44]. It is extremely difficult to estimate the concentrations of these constituents in vivo as very little is known about disposition of flavonoids in humans (46). A previous study has demonstrated that the grapefruit flavonoids naringin and hesperidin are absorbed from the human gastrointestinal tract after oral administration of pure compounds, but their oral bioavailability was very low, i.e. < 20% (46). The contribution of each component varies according to its inhibitory potency and natural abundance. Thus, the in vivo effect of grapefruit juice is obviously complicated.
Naringin was originally thought to be a possible active component. However, as mentioned earlier, when an aqueous solution of naringin was given to volunteers, it produced little or no interaction with felodipine (14). A recent study concluded that naringin by itself at concentrations found in grapefruit juice does not appear to be capable of producing a clinical drug interaction such as that seen with grapefruit juice (47). The furanocoumarins, bergamottin, 6',7'-dihydroxybergamottin, and related dimers (25,47,48) seem to be more important active ingredients involved in the interaction, although other unidentified substances may be also involved in this clinical interaction. Being a potent inhibitor of CYP3A4, bergapten is likely to be an additional active ingredient in grapefruit juice-drug interactions.
In summary, the present study has demonstrated that besides the flavonoids, other compounds found in grapefruit including furanocoumarins can produce strong inhibition of CYP3A4. The grapefruit juice-drug interactions could involve CYP3A4 inhibition by more than one component present in grapefruit juice. Bergapten was found to be a very potent inhibitor of CYP3A4. Therefore, it may be an important furanocoumarin responsible for the grapefruit juice drug interactions. Concentrations of bergapten (up to 30 mM) detected in grapefruit and commercial grapefruit juice products (44) appear sufficient to cause significant inhibition of CYP3A4 in this study. However, it should be noted that inhibition of CYP3A4 activity in vitro does not necessarily imply drug interaction in vivo . Further studies will be needed to determine if this furanocoumarin bergapten can influence the CYP enzyme in vivo .
Acknowledgments
This study was supported by a research grant from the Health Research Council (HRC) of New Zealand.
References
Hertog, M.G.L., Feskens, E.J.M., Hollman, P.C.H.,Katan, M.B. and Kromhout, D., Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen elderly study. Lancet, 342:1007-1011, 1993.
Conney, A.H., Buening, M.K., Pantuck, E.J., Fortner, J.G. and Anderson, K.E., Regulation of human drug metabolism by dietary factors, in: Evered, D. and Lawrenson, G. (eds.), Environmental Chemicals, Enzyme Function and Human Disease. Excepta Medica, Amsterdam, pp 147-167, 1980.
Bailey, D.G, Arnold, J.M.O. and Spence, J.D., Grapefruit juice-drug interactions. Br J Clin Pharmacol, 46:101-110, 1998.
Kolars, J.C., Awni W.M., Merion, R.M. and Watkins, P.B., First-pass metabolism of cyclosporin by the gut. Lancet, 338:1488-1490, 1991.
Lown, K.S., Bailey, D.G., Fontana, R.J., Janardan, S.K., Adair, C.H., Fortlage, L.A., Brown, M.B., Guo, W. and Watkins, P.B., Grapefruit juice increases felodipine oral availability in humans by decreasing intestinal CYP3A protein expression. J Clin Invest, 99:2545-2553, 1997.
Paine, M.F., Shen, D.D., Kunze, K.L., Perkins, J.D., Marsh, C.L., McVicar, J.P., Barr, D.M., Gillies, B.S. and Thummel, K.E., First-pass metabolism of midazolam by the human intestine. Clin Pharmacol Ther, 60:14-24, 1996.
Guengerich, F.P., Human Cytochrome P450 Enzymes, in: Ortiz de Montelano P.R., (ed.), Cytochrome P450-Structure, Mechanism and Biochemistry, 2nd ed., Plenum Press, New York, pp 473-535, 1995.
Watkins, P.B., Wrighton, S.A., Schuetz, E.G., Molowa, D.T. and Guzelian, P.S., Identification of glucocorticoid-inducible cytochromes P-450 in the intestinal mucosa of rats and man. J Clin Invest, 80:1029-1036, 1987.
Fuhr, U., Klittich, K., Staib, A.H., Inhibitory effect of grapefruit juice and its bitter principal, naringenin, on CYP1A2 dependent metabolism of caffeine in man. Br J Clin Pharmacol, 35: 431436, 1993.
Merkel, U., Sigusch, H., Hoffmann, A., Grapefruit juice inhibits 7-hydroxylation of coumarin in healthy volunteers. Eur J Clin Pharmacol, 46: 175177, 1994.
Runkel, M., Bourian, M., Tegtmeier, M., Legrum, W., The character of inhibition of the metabolism of 1,2-benzopyrone (coumarin) by grapefruit juice in human. Eur J Clin Pharmacol, 53: 265269, 1997.
Guengerich, F.P. and Kim, D.H., In vitro inhibition of dihydropyridine oxidation and aflatoxin B1 activation in human liver microsomes by naringenin and other flavonoids. Carcinogenesis, 11: 2275-2279, 1990.
Miniscalco, A., Lundahl, J., Regårdh, C.G., Edgar, B. and Eriksson, U.G., Inhibition of dihydropyridine metabolism in rat and human liver microsomes by flavonoids found in grapefruit juice. J Pharmacol Exp Ther, 261:1195-1199, 1992.
Bailey, D.G., Arnold, J.M.O., Munoz, C. and Spence, J.D., Grapefruit juice-felodipine interaction: mechanism, predictability, and effect of naringin. Clin Pharmacol Ther, 53:637-642, 1993.
Bailey, D.G., Arnold, J.M.O., Strong, H.A., Munoz, C. and Spence, J.D., Effect of grapefruit juice and naringin on nisoldipine pharmacokinetics. Clin Pharmacol Ther, 54:589-594, 1993.
Rashid, J., Mckinstry, C., Renwick, A.G., Dirnhuber, M., Waller, D.G. and George, C.F., Quercetin, an in vitro inhibitor of CYP3A4, does not contribute to the interaction between nifedipine and grapefruit juice. Br J Clin Pharmacol, 36:460-463, 1993.
Edwards, D.J., Bellevue, F.H. and Woster, P.M., Identification of 6',7'-dihydroxybergamottin, a cytochrome P450 inhibitor in grapefruit juice. Drug Metab Dispos, 24:128-129, 1996.
Fukuda, K., Ohta, T. and Yamazoe, Y., Grapefruit component interacting with rat and human P450 CYP3A: possible involvement of non-flavonoid components in drug interaction. Biol Pharm Bull, 20: 560-564, 1997.
He, K., Iyer, K.R., Hayes, R.N., Sinz, M.W., Woolf, T.F. and Hollenberg, P.F., Inactivation of cytochrome P450 3A4 by bergamottin, a component of grapefruit juice. Chem Res Toxicol, 11:252-259, 1998.
Zhang, H., Coville, P.F., Walker, R.J., Miners, J.O., Birkett, D.J. and Wanwimolruk, S., Evidence for involvement of human CYP3A in the 3-hydroxylation of quinine. Br J Clin Pharmacol, 43:245-252, 1997.
Lown, K.S., Ghosh, M, Watkins, P.B., Sequences of intestinal and hepatic cytochrome P450 3A4 cDNAs are identical. Drug Metab Dispos, 26:185-7, 1998.
Ha, H.R., Chen, J., Leuenberger, P.M., Freiburghaus, A.U., Follath, F., In vitro inhibition of midazolam and quinidine metabolism by flavonoids. Eur J Clin Pharmacol;48:367-71, 1995.
Obermeier, M.T., White, R.E. and Yang, C.S., Effects of bioflavonoids on hepatic P450 activities. Xenobiotica, 25:575-584, 1995.
Wanwimolruk S, Wong SM, Zhang H and Coville PF., Simultaneous determination of quinine and a major metabolite 3-hydroxyquinine in biological fluids by HPLC without extraction. J Liq Chromatogr Rel Technol, 19:293-305, 1996.
Guo, L.Q., Fukuda, K. Ohta, T. and Yamazoe, Y., Role of furanocoumarin derivatives on grapefruit juice-mediated inhibition of human CYP3A activity. Drug Metab Dispos, 28: 766-771, 2000.
Lasker, J.M., Huang, M.T. and Conney, A.H., In vitro and in vivo activation of oxidative drug metabolism by flavonoids. J Pharmacol Exp Ther, 229:162-170, 1984.
Siess, M., Leclerc J., Canivenc-Lavier, Rat P. and Suschetet M., Heterogenous effects of natural flavonoids on monooxygenase activities in human and rat liver microsomes. Toxicol Appl Pharmacol, 130:73-78, 1995.
Beyeler, S., Testa, B. and Perrissoud, D., Flavonoids as inhibitors of rat liver monooxygenase activities. Biochem Pharmacol, 37: 1971-1979, 1988.
Li, Y., Wang, E., Patten, C.J., Chen, L. and Yang, C.S., Effects of flavonoids on cytochrome P450 dependent acetaminophen metabolism in rats and human liver microsomes. Drug Metab Dispos, 22:566-571, 1994.
Bors, W., Heller, W., Michel, C. and Saran, M., Flavonoids as antioxidants: determination of radical-scavenging. Methods Enzymol, 186:343-355, 1990.
Zhang, Y.D., Lorenzo, B. and Reidenberg, M.M., Inhibition of 11β-hydroxysteroid dehydrogenase obtained from guinea pig kidney by furosemide, naringenin and some other compounds. J Steroid Biochem Mol Biol, 49:81-5, 1994.
Lee, Y.S., Lorenzo, B.J., Koufis, T. and Reidenberg, M.M., Grapefruit juice and its flavonoids inhibit 11b-hydroxysteroid dehydrogenase. Clin Pharmacol Ther, 59:62-71, 1996.
Buening, M.K., Chang, R.L., Huang, M.T., Fortner, J.G., Wood, A.W. and Conney, A.H., Activation and inhibition of benzo(a)pyrene and aflatoxin B1 metabolism in human liver microsomes by naturally occurring flavonoids. Cancer Res, 41: 67-72, 1981.
Stanley, W.L. and Leonard, J., Citrus coumarins. J Agr Food Chem, 19: 1106-1110, 1971.
Beier, R.C., Natural pesticides and bioactive components in foods. Rev Environ Contam Toxicol, 113:47-137, 1990.
Schmiedlin-Ren, P., Edwards, D.J., Fitzsimmons, M.E., He, K., Lown, K.S., Woster, P.M., Thummel, K.E., Fisher, J.M., Hollenberg, P.F. and Watkins, P.B. Mechanism of enhanced oral availability of CYP3A4 substrates by grapefruit constituents- decreased enterocyte CYP3A4 concentration and mechanism-based inactivation by furanocoumarins. Drug Metab Dispos, 25:1228-1233, 1997.
Bellevue, F.H., Woster, P.M., Edwards, D.J., He, K. and Hollenberg, P.F., Synthesis and biological evaluation of 6',7'-dihydroxybergamottin (6',7'-DHB), a naturally occurring inhibitor of cytochrome P450 3A4. Bioorg. Med Chem Lett, 7:2593-2598, 1997.
Ueng, Y.F., Kuwabara, T., Chun, Y.J. and Guengerich, F.P., Cooperativity in oxidations catalyzed by cytochrome P4503A4. Biochemistry, 36:370-381, 1997.
Malhotra, S., Bailey, D.G., Paine, M.F. and Watkins, P.B., Seville orange juice-felodipine interaction: comparison with dilute grapefruit juice and involvement of furocoumarins. Clin Pharmacol Ther, 69:14-23, 2001.
Cai,Y., Baer-Dubowska,W., Ashwood-Smith, M.J., Ceska, O., Tachibana, S. and DiGiovanni, J., Mechanism-based inactivation of hepatic ethoxyresorufin O-dealkylation activity by naturally occurring coumarins. Chem Res Toxicol, 9: 729-736, 1996.
Tinel, M., Belghiti, J., Descatoire, V., Amouyal, G., Letteron, P., Geneve, J., Larrey, and D.,Pessayre, D., Inactivation of human liver cytochrome P-450 by the drug methoxalen and other psoralen derivatives. Biochem Pharmacol, 36: 951-955, 1987.
Commandeur, J.N.M. and Vermeulen, N.P.E., Molecular and biochemical mechanisms of chemically induced nephrotoxicity: a review. Chem Res Toxicol, 3: 485-491, 1990.
Fouin-Fortunet, H., Tinel, M., Descatoire, V., Lettern, P., Lerrey, D., Geneve, J. and Pessayre, D., Inactivation of cytochrome P-450 by the drug methoxalen. J Pharmacol Exp Ther, 236: 237-247, 1986.
Ho, P.C., Saville, D.J., Coville, P.C. and Wanwimolruk, S., Content of CYP3A4 inhibitors, naringin, naringenin and bergapten in grapefruit and grapefruit juice products. Pharmaceut Acta Helv, 74:379-385, 2000.
Ruseff, R.L., Martin, S.F., Youtsey, C.O., Quantitative survey of narirutin, naringin, hesperidin and neohesperidin in Citrus. J Agric Food Chem, 35:1027-1030, 1987.
Ameer, B,, Weintraub, R.A., Johnson, J.V., Yost, R.A., Rouseff, R.L., Flavanone absorption after naringin, hesperidin, and citrus administration. Clin Pharmacol Ther, 60:34-40, 1996.
Bailey, D.G., Dresser, G.K., Kreeft, J.H., Munoz, G., Freeman, D.J. and Bend, J.R., Grapefruit juice-felodipine interaction: effect of unprocessed fruit and probable active ingredients. Clin Pharmacol Ther, 68:468-477, 2000.
Kukuda, K., Ohta, T., Oshima, Y., Ohashi, N., Yoshikawa, M. and Yamazoe, Y., Specific CYP3A4 inhibitors in grapefruit juice: furanocoumarin dimers as components of drug interaction. Pharmacogenetics, 7:391-396, 1997.
Corresponding Author: Sompon Wanwimolruk, College of Pharmacy, Western University of Health Sciences, 309 E. Second Street, Pomona, California, USA, 91766-1854. swanwimolruk@westernu.edu
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps