J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 4(3):263-268, 2001
High-performance liquid chromatographic assay of amiodarone in rat plasma
Andrew S. Jun, Dion R. Brocks1
College of Pharmacy, Western University of Health Sciences, California, USAManuscript received October 17, 2001, Revised November 27, Accepted November 28, 2001
PDF Version
Abstract
Purpose. A high-performance liquid chromatographic method is described for the determination of amiodarone in rat plasma.
Methods. After liquid-liquid extraction, the separation of amiodarone from internal standard and endogenous components was accomplished using reversed phase chromatography. The mobile phase, a combination of monobasic potassium phosphate, methanol and acetonitrile, was run isocratically through a C8 analytical column. The UV detection was at 254 nm for ethopropazine, the internal standard, and subsequently changed to 242 nm for amiodarone detection.
Results. Analytical run time was less than 13 min. Mean recovery was 75% and 82% for lower (0.5 mg/ml) and higher concentrations (5 mg/ml), respectively. The assay exhibited excellent linear relationships between peak height ratios and plasma concentrations; quantitation limit was at least 0.035 mg/ml, based on 100 ml of rat plasma. Accuracy and precision were <17% over the concentration range of 0.035 to 5 mg/ml.
Conclusion. The assay was applied successfully to the measurement of amiodarone plasma concentrations in rats given the drug orally.
Introduction
Amiodarone [2-butyl-3-(3,5-diiodo-4b-diethylaminoethoxybenxoyl) benzofuran] is a class III antiarrhythmic agent used to treat patients afflicted with a number of arrhythmias of atrial or ventricular origin (1). In recent years, amiodarone has become an increasingly useful and prescribed antiarrhythmic drug. This increase in amiodarone use has been prompted in large part by the realization that alternative antiarrhythmic drugs, such as the class IC agent, encainide, are prone to increase mortality in post-myocardial infarction patients (2).
Amiodarone possesses an interesting pharmacokinetic profile, with long elimination half-life, large volume of distribution, and erratic and sometimes incomplete bioavailability (3, 4). A number of analytical assays for use in pharmacokinetic studies are available for determination of amiodarone in biological specimens (Table 1).
Table 1: Previously published assays for amiodarone in blood fluids.
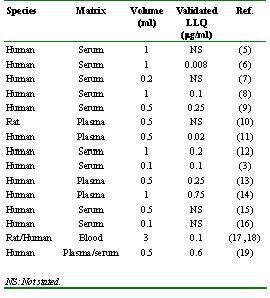
Most of these assays (3, 5-19) are intended for use in human plasma, and typically require specimen volumes ranging from 0.5 to 1 mL of plasma (Table 1). Rat is a commonly used animal model, and pharmacokinetic studies of amiodarone in rats have mostly been non-survival in nature (17, 20, 21), involving one animal per specimen concentration, with the need for relatively large volumes (0.5 to 3 ml) of blood, serum or plasma per individual sample. Volumes of this size are too large to permit serial blood sample collection in survival, non-terminal pharmacokinetic and pharmacodynamic studies.
Here we describe a high-performance liquid chromatographic (HPLC) method for assay of amiodarone in small volumes of rat plasma. The method can be used in pharmacokinetic studies of amiodarone where multiple, serial blood sample collection is desired in individual rats.
Experimental
Materials and chemicals
Amiodarone HCl and ethopropazine HCl were obtained from Sigma (St. Louis, MO, USA). Methanol, acetonitrile, hexane (all HPLC grades), triethylamine, potassium phosphate (monobasic), sodium phosphate (mono and dibasic) and sulfuric acid were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Water for analytical purposes was obtained using a Barnstead NanoPure Infinity water purification system (Dubuque, IA, USA).
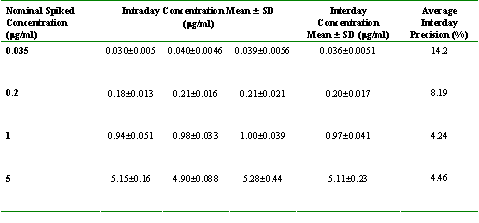
Table 2: Validation data for assay of amiodarone in rat plasma.
Chromatographic conditions
The HPLC system consisted of a Shidmadzu LC-10AT liquid chromatograph pump, SIL-10A autosampler and SPD-10A UV-VIS UV absorbance detector (Shimadzu, Kyoto, Japan). Data collection, integration and calibration were accomplished using Class VP Chromatography Data System Version 4.2 computer software (Shimadzu, Kyoto, Japan).
The chromatographic separations of amiodarone and internal standard (ethopropazine) were accomplished using a 100 mm × 8 mm I.D. Nova-Pak C8 analytical column cartridge placed in a radial compression module (Waters, Milford MA, USA). A Guard-Pak Precolumn Module (Waters, Milford MA, USA) containing an ODS cartridge insert was placed serially just before the analytical column. The mobile phase consisted of methanol: acetonitrile: [25 mM KH2 PO4:3 mM sulfuric acid: 3.6 mM triethylamine] in a combination of 63:12:25 v/v. Before use, the mobile phase was degassed by passing it through a 0.45 mm filter.
The mobile was pumped at an isocratic flow rate of 1.5 ml/min at room temperature. Immediately after injection of the sample into the HPLC, the UV detection wavelength was set at 254 nm. At 7 min post-injection, it was switched to 242 nm. The wavelengths of 254 and 242 nm represented the UV maximum of ethopropazine and amiodarone, respectively, in methanol.
Standard and stock solutions
A stock drug solution was prepared by dissolving 3.4 mg of amiodarone HCl in 32.2 ml of methanol, representing 100 mg/ml of amiodarone base. This solution was stored at -20°C between use; amiodarone is known to remain stable for at least 3 months under these conditions (5).
The working standard solutions were prepared daily from the stock solution by sequential dilution with methanol to yield final concentrations of 10, 1 and 0.1 mg/ml amiodarone. The internal standard stock solutions were prepared by dissolving 5 mg of ethopropazine HCl in 100 ml of methanol (50 mg/ml). This solution was stored at -20°C between use.
Extraction procedure
In a 1.5-ml polypropylene centrifuge tube, 100 ml of rat plasma was added along with 30 ml of internal standard solution. Plasma proteins were precipitated by the addition of 300 ml of acetonitrile, and then the tubes were vortex mixed. The tubes were subsequently centrifuged for 2 min and the supernatant was carefully transferred to new glass tubes using clean Pasteur pipets. To each tube, 300 ml of phosphate buffer (pH = 5.9) and 3 ml of hexane were added. The tubes were then vortex mixed for 30 s and centrifuged at 3000 g for 3 min. The organic solvent layer was transferred to new tubes and evaporated to dryness in vacuo. The residues were reconstituted using 150 ml of mobile phase and aliquots of 30 ml were injected into the HPLC.
Recovery
The extraction efficiency was determined by comparing the peak heights of known amounts of amiodarone (unextracted) in mobile phase directly injected to peak heights of samples containing the same amounts of amiodarone in plasma after extraction. Recovery was determined at 0.5 mg/ml (n=4) and 5 mg/ml (n=4) of amiodarone.
Calibration, accuracy and precision
Quantification was based on calibration curves constructed using peak height ratios of drug to internal standard vs. nominal drug concentration. Intra-day reproducibility was tested by using four different concentrations per day in quadruplicate (0.035, 0.2, 1, and 5 mg/ml). The procedure was repeated on three separate days to allow for determination of inter-day precision and accuracy. Intra-day accuracy was estimated based on the mean percentage error, and the inter-day accuracy was calculated as the mean of the intra-day accuracy determinations. The precision, expressed as a percentage, was evaluated by calculating the intra- and inter-day relative standard deviations.
Animal study
In order to assess the ability of the assay to measure amiodarone concentrations in vivo samples, a preliminary pharmacokinetic study was conducted in two male Sprague-Dawley rats (350-450 g each). Under anesthesia with halothane administered using an anaesthetic machine, a Micro-renathane cannula (Braintree scientific, Braintree, MA) was implanted into the right jugular vein of each animal. One rat received a single 20 mg/kg oral dose of amiodarone in 1% methylcellulose suspension, whereas the other received 25 mg/kg amiodarone HCl as intravenous bolus. For intravenous dosing, the amiodarone HCl was dissolved in a mixture of acetic acid: N,N-dimethylacetamide: polyethylene glycol 400: 5% dextrose in water (1:3:17:30) to provide for a amiodarone HCl dosing solution of 10 mg/mL. Serial blood samples were collected from the cannula for 48 h, and the resultant plasma samples were kept at -20°C until assayed. The area under the plasma concentration vs. time curve from time of dosing to last plasma sample (AUC0-48h ) was determined using the log-linear trapezoidal rule.
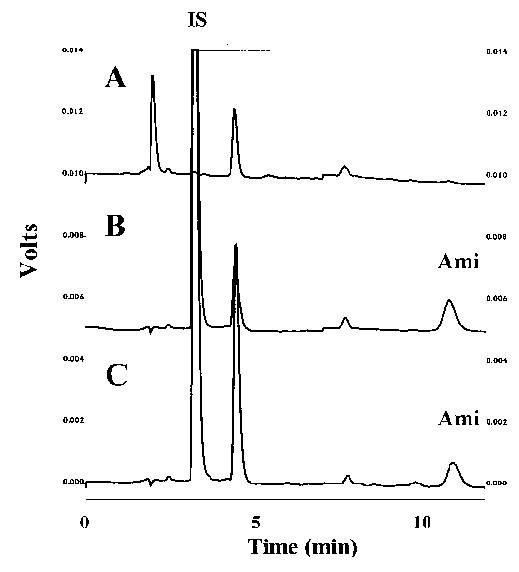
Figure 1: Chromatograms derived from assay of A.) blank rat plasma, B.) rat plasma spiked with 0.5
mg/mL amiodarone (AMI) and C.) rat plasma 3 h after oral administration of 20 mg/kg amiodarone HCl. IS denotes the internal standard (ethopropazine).
Results
Peaks corresponding to internal standard and amiodarone eluted free of interfering substances, at 3.2 and 10.8 min, respectively (Fig 1).
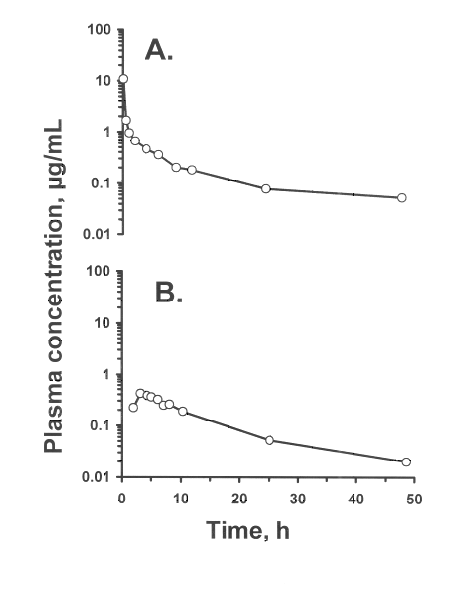
Figure 2: Plasma amiodarone concentration vs. time curves from rats dosed with A.) 25 mg/kg intravenous amiodarone HCl, and B.) 20 mg/kg amiodarone HCl via oral gavage.
The analytical run time was 13 min for each plasma sample. The mean extraction efficiencies of amiodarone from 100 ml of rat plasma, at concentrations of 0.5 and 5 mg/ml, were 75.0% and 82.1%, respectively. For construction of calibration curves, the data were weighted by the factor 1/concentration. Excellent linear relationships (r2≥0.994) were obtained between peak height ratios and the corresponding plasma concentrations over a range of 0.035 to 50 mg/ml of amiodarone. A typical regression line from the validation run was described by amiodarone concentration ( mg/ml) = (peak height ratio + 0.00408)/12.825.
The intraday and interday coefficients of variation were less than 17% (Table 2). Over the range of concentrations from 0.035 to 5 mg/ml, the intraday accuracies ranged from 84.8 to 114%, and average interday accuracy ranged from 97.4 to 103% (Table 2). Based on this data, the validated lower limit of quantitation of the method was 0.035 mg/ml based on 100 ml of rat plasma (Table 2).
In the rat given a 20 mg/kg oral dose of amiodarone, plasma samples were analyzed using the described HPLC method (Fig 2). In this rat, the maximum plasma concentration was 0.42 ng/mL at 3 h postdose. The area under the plasma concentration-time curve (AUC0-48h ) was 5.4 mg·h/l. In the rat given 25 mg/kg amiodarone HCl intravenously, the AUC0-48h was 9.4 mg·h/l.
Discussion
The HPLC method described here was accurate and precise, and capable of determining concentrations of amiodarone in small volumes of rat plasma. The extraction procedure was simple and the procedure used a commercially available internal standard (ethopropazine). The method performed well with respect to reproducibility and accuracy over the range of concentrations studied (Tables 1). This assay method was rapid; preparation of 30 samples took less than 2 h from initial protein precipitation to final placement of samples in the HPLC autosampler vials. The chromatographic run time was less than 13 min. The lower limits of quantitation and the small plasma sample necessary for this assay was ideal for studying amiodarone pharmacokinetics in the rat.
Other investigators have described analytical methods for the determination of amiodarone in human plasma, (Table I). However, these methods require the use of relatively large volumes of plasma (0.5 to 1.0 ml) and the reported validated lower limits of quantitation are typically greater than 0.1 mg/ml. The lowest validated quantifiable concentration we could find in the literature was 0.008 mg/ml, although a plasma volume of 1 mL was required (6). Some of the reported assays require normal phase chromatography with volatile solvents, which can be disadvantageous from the perspective of laboratory safety and occupational health. The assay described here uses reverse phase chromatography, which is less burdened by such issues.
A few procedures have been reported for assay of amiodarone in rat (10, 17). In most existing pharmacokinetic studies involving rats, animals were sacrificed at set times after dosing with drug, and relatively large volumes of plasma or blood were assayed for measurement of drug content (17, 20, 21). For survival studies, serum or plasma volumes of 0.5-1.5 ml, representing 1-3 ml of blood, are too large for repeated sampling from rats. The present assay permits use in non-terminal pharmacokinetic studies owing to its smaller sample volume (0.1 ml). In addition, the assay is very sensitive, permitting reliable measurement of amiodarione concentrations as low as 0.035 mg/ml.
Some assay procedures are capable of simultaneously measuring desethylamiodarone in plasma. We could not test the ability of our assay to measure this metabolite due to the unavailability of the preformed metabolite. Due to the increased hydrophilicity of the metabolite, it would be expected to elute at an earlier time than amiodarone under the reversed phase conditions used here. We did observe the presence of a peak in the chromatograms at 9.9 min in the in vivo rat samples. Because this peak appeared to grow in size as time progressed, it is possible that this peak represented desethylamiodarone or some other more polar metabolite of amiodarone.
In conclusion, we have described an analytical method capable of measuring amiodarone in small volumes of plasma. The assay was found to be suitable for pharmacokinetic studies in rats given oral doses of amiodarone, and was validated for measurement of concentrations as low as 35 ng/mL of amiodarone based on 100 ml of rat plasma.
Presented in part at the American Association of Pharmaceutical Scientists 2001 Annual Meeting and Exposition, October 21-25, Denver, Colorado, USA.
References
Jafari-Fesharaki M. and Scheinman M.M., Adverse effects of amiodarone. Pacing Clin Electrophysiol, 21: 108-20, 1998.
Lazzara R., From first class to third class: recent upheaval in antiarrhythmic therapy--lessons from clinical trials. Am J Cardiol, 78: 28-33, 1996.
Pollak P.T.;Bouillon T. and Shafer S.L., Population pharmacokinetics of long-term oral amiodarone therapy. Clin Pharmacol Ther, 67: 642-52, 2000.
Meng X.;Mojaverian P.;Doedee M.;Lin E.;Weinryb I.;Chiang S.T. and Kowey P.R., Bioavailability of amiodarone tablets administered with and without food in healthy subjects. Am J Cardiol, 87: 432-5, 2001.
Ress K.;Liebich H.M.;Kramer B.;Ickrath O.;Risler T. and Seipel L., Determination of amiodarone and its metabolite N-desethylamiodarone in serum by high-performance liquid chromatography--comparison of different extraction procedures. J Chromatogr, 417: 465-70, 1987.
Bliss M.;Mayersohn M. and Nolan P., High-performance liquid chromatographic analysis of amiodarone and desethylamiodarone in serum. J Chromatogr, 381: 179-84, 1986.
Kannan R.;Miller S.;Perez V. and Singh B.N., Sensitive method for the measurement of amiodarone and desethylamiodarone in serum and tissue and its application to disposition studies. J Chromatogr, 385: 225-32, 1987.
Brien J.F.;Jimmo S. and Armstrong P.W., Rapid high-performance liquid chromatographic analysis of amiodarone and N-desethyl-amiodarone in serum. Can J Physiol Pharmacol, 61: 245-8, 1983.
Lesko L.J.;Marion A.;Canada A.T. and Haffajee C., High-pressure liquid chromatography of amiodarone in biological fluids. J Pharm Sci, 70: 1366-8, 1981.
Moor M.J.;Wyss P.A. and Bickel M.H., New procedure for the high-performance liquid chromatographic determination of amiodarone and desethylamiodarone with solid-phase extraction of rat plasma and tissue samples. J Chromatogr, 431: 455-60, 1988.
Hutchings A.;Spragg B.P. and Routledge P.A., High-performance liquid chromatographic assay of amiodarone and desethylamiodarone in plasma. J Chromatogr, 382: 389-93, 1986.
Muir K.T.;Kook K.A.;Stern C. and Gardner K.M., Analysis of amiodarone and desethylamiodarone in serum and tears by reversed-phase high-performance liquid chromatography. J Chromatogr, 374: 394-9, 1986.
Duranti L.;Caracciolo M. and Oriani G., New, accurate semi-automatic high-performance liquid chromatographic method for routine monitoring of amiodarone plasma levels. J Chromatogr, 277: 401-7, 1983.
Plomp T.A.;Engels M.;Robles de Medina E.O. and Maes R.A., Simultaneous determination of amiodarone and its major metabolite desethylamiodarone in plasma, urine and tissues by high-performance liquid chromatography. J Chromatogr, 273: 379-92, 1983.
Mostow N.D.;Noon D.L.;Myers C.M.;Rakita L. and Blumer J.L., Determination of amiodarone and its N-deethylated metabolite in serum by high-performance liquid chromatography. J Chromatogr, 277: 229-37, 1983.
Lam S., High-performance liquid chromatography of amiodarone and desethylamiodarone in serum after microscale protein precipitation. J Chromatogr, 381: 175-8, 1986.
Riva E.;Gerna M.;Neyroz P.;Urso R.;Bartosek I. and Guaitani A., Pharmacokinetics of amiodarone in rats. J Cardiovasc Pharmacol, 4: 270-5, 1982.
Riva E.;Gerna M.;Latini R.;Giani P.;Volpi A. and Maggioni A., Pharmacokinetics of amiodarone in man. J Cardiovasc Pharmacol, 4: 264-9, 1982.
Lesne M. and Pellegrin P.L., Rapid high-performance liquid chromatographic method for the determination of amiodarone and desethylamiodarone in human plasma and serum. J Chromatogr, 415: 197-202, 1987.
Plomp T.A.;Wiersinga W.M. and Maes R.A., Tissue distribution of amiodarone and desethylamiodarone in rats after multiple intraperitoneal administration of various amiodarone dosages. Arzneimittelforschung, 35: 122-9, 1985.
Najjar T.A., Disposition of amiodarone in rats after single and multiple intra-peritoneal doses. Eur J Drug Metab Pharmacokinet, 25: 199-203, 2000.
Corresponding Author: Dion R. Brocks, Associate Professor of Pharmaceutical Sciences, Western University of Health Sciences, College of Pharmacy, 309 E. Second Street/College Plaza, Pomona, California, USA, 91766-1854. dionbrocks@yahoo.com
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps
