J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 4(2):185-200, 2001
Stereospecific Pharmacokinetics and Pharmacodynamics of Beta-Adrenergic Blockers in Humans
Reza Mehvar1
School of Pharmacy, Texas Tech University Health Sciences Center, Amarillo, Texas, USADion R. Brocks
College of Pharmacy, Western University of Health Sciences, Pomona, California, USAReceived May 18th, 2001, Revised July 5th, 2001, Accepted July 5th, 2001
PDF version
Abstract
The beta-blockers comprise a group of drugs that are mostly used to treat cardiovascular disorders such as hypertension, cardiac arrhythmia, or ischemic heart disease. Each of these drugs possesses at least one chiral center, and an inherent high degree of enantioselectivity in binding to the b-adrenergic receptor. For beta-blockers with a single chiral center, the (-) enantiomer possesses much greater affinity for binding to the b-adrenergic receptors than antipode. The enantiomers of some of these drugs possess other effects, such as antagonism at alpha-adrenergic receptors or Class III antiarrhythmic activity. However, these effects generally display a lower level of stereoselectivity than the beta-blocking activity. Except for timolol, all of these drugs used systemically are administered clinically as the racemate. As a class, the beta blockers are quite diverse from a pharmacokinetic perspective, as they display a high range of values in plasma protein binding, percent of drug eliminated by metabolism or unchanged in the urine, and in hepatic extraction ratio. With respect to plasma concentrations attained after oral or intravenous dosing, in most cases the enantiomers of the beta-blockers show only a modest degree of stereoselectivity. However, the relative magnitude of the concentrations of the enantiomers in plasma is not constant in all situations and varies from drug to drug. Further, various factors related to the drug (e.g., dosing rate or enantiomer-enantiomer interaction) or the patient (e.g., racial background, cardiovascular function, or the patient metabolic phenotype) may affect the stereospecific pharmacokinetics and pharmacodynamics of beta-blockers. An understanding of the stereospecific pharmacokinetics and pharmacodynamics of beta-blockers may help clinicians to interpret and predict differences among patients in pharmacologic responses to these drugs.
Introduction
In clinical practice, the b-adrenergic antagonists are an extremely important class of drugs due to their high prevalence of use. Many have been synthesized and are commonly used systemically in the treatment of conditions including hypertension, cardiac arrhythmia, angina pectoris, and acute anxiety, and topically for open angle glaucoma. With respect to their clinical utility, the beta-blockers are normally distinguished based on their selectivity for beta-receptors. The nonselective beta-blockers, including propranolol, oxprenolol, pindolol, nadolol, timolol and labetalol, each antagonize both b1 - and b2 -adrenergic receptors. For the selective antagonists, including metoprolol, atenolol, esmolol, and acebutolol, each has much greater binding affinity for the b1 adrenergic receptor. The selective beta-blockers are normally indicated for patients in whom b2 -receptor antagonism might be associated with an increased risk of adverse effects. Such patients include those with asthma or diabetes, or patients with peripheral vascular disease or Raynaud's disease.
As depicted in Figure 1, a common feature in the chemical structure of beta-blockers is that there is at least one aromatic ring structure attached to a side alkyl chain possessing a secondary hydroxyl and amine functional group. Each of the available beta-blockers has one or more chiral centers in its structure, and in all cases, at least one of the chiral carbon atoms residing in the alkyl side chain is directly attached to a hydroxyl group. Except for timolol, which is marketed as S-enantiomer, each of the beta-blockers with one chiral center (e.g., propranolol, metoprolol, atenolol, esmolol, pindolol, and acebutolol) is marketed as a racemate consisting of two enantiomers. Additionally, labetalol, which has two chiral centers (Figure 1), is marketed as a racemate consisting of four isomers. As for nadolol, the drug has three chiral centers in its structure (Figure 1). However, the two ring hydroxyl groups (Figure 1) are in the cis orientation, allowing for only four isomers.
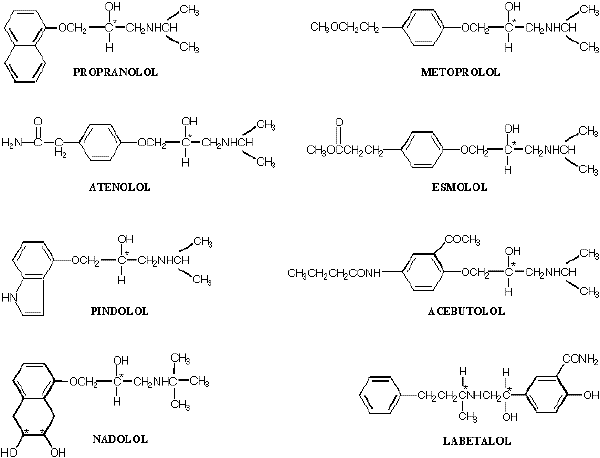
Figure 1. Chemical structure of major beta-adrenoceptor blocking agents. The asterisk denotes the chiral carbon.
Amongst chiral drugs, arguably the beta-adrenergic blocking drugs are one of the best-understood classes from the perspective of stereoselectivity in pharmacokinetics and pharmacodynamics. The enantiomers of b-blockers possess markedly different pharmacodynamics, and in some cases, pharmacokinetics. Although there is no defined range of plasma concentrations for the beta-blockers, for some of them, an effective concentration has been proposed (1). The intent of this review is to summarize what is known of the pharmacodynamic and pharmacokinetic properties of some of the major b-adrenergic antagonists in current use.
Stereoselectivity in Pharmacologic Action
The available data regarding the pharmacologic action of beta-blockers indicate that the interaction of these agents with beta-adrenoceptors is highly stereoselective (2-12). Generally, the cardiac beta-blocking activity of the beta-blockers with two enantiomers resides in their S (-) enantiomers (2-7), the reported S:R activity ratio being in the range of 33 to 530 (Table 1). However, the R (+)-enantiomer has relatively strong activity in blocking b2 receptors in ciliary process (4) (Table 1). For sotalol, which has R (-) and S (+) conformation, it is the R (-) enantiomer that possesses the majority of the b-blocking activity (8). Both enantiomers of sotalol share an equivalent degree of Class III antiarrhythmic potency, however (8).
Table 1. Stereoselectivity in the action of Some beta-adrenergic blockers.
Drug
Relative Activity (Ratio)
Experimental Model
Biological Response
Reference
Atenolol
> +
Rat
Reduction in heart rate and mean arterial pressure
(2)
> + (46:1)
Guinea pig
Beta-blocking activity of heart
(3)
Betaxolol
> + (530:1)
Rabbit
Beta-blocking activity of heart
(4)
+ > (190:1)
Blocking b2 receptors in ciliary process
Bucindolol
> +
Dog
Myocardial stimulant, vasodilator
(5)
Carvedilol
> +
Rat and Rabbit
Beta-blocking activity
(14)
= +
Alpha-blocking activity
Labetalol
RR > SR; RR > RS; RR > SS
Rat
Beta-blocking activity
(10)
SR > RR; SR > RS; SR > SS
Alpha-blocking activity
RR-SR > labetalol (2:1)
Rat
Antagonizing pressor effects of phenylephrine and chronotropic effect of isoprenaline
(11)
RR-SR > labetalol
Alpha1-blocking activity
RR-SR > labetalol (3:1)
Beta1-blocking activity
Metoprolol
> + (33:1)
Rabbit
Beta-blocking activity of heart
(4)
+ > (10:1)
Blocking b2 receptors in ciliary process
Nadolol
SQ-12151 > SQ-12150 > nadolol > SQ-12148 > SQ-12149
Chinese hamster ovary cells
Binding values to b1, b2, and b3 cloned receptors
(12)
Pindolol
> + (200:1)
Guinea pig
Blocking b1 and b2 receptors
(6)
Propranolol
> + (100:1)
Rat
Frog nerveBlocking isoprenaline cardiac response
(7)
= +
Local anesthetic effects
Sotalol
> +
Dog and rabbit
Beta-blocking activity
(8)
= +
Class III antiarrhythmic activity
Timolol
> + (44:1)
Rabbit
Beta-blocking activity of heart
(4)
= +
Blocking b2 receptors in ciliary process
Carvedilol is a newer agent that is marketed as the racemate for the treatment of hypertension and congestive heart failure (13). This latter indication is unique amongst the available beta-blockers, for which this condition is normally a contraindication. Similar to other beta-blockers, the S (-) enantiomer of carvedilol is more potent as an antagonist of beta-receptors. Both the R and S enantiomers, however, are equally effective in blocking alpha-adrenergic activity (14). This gives the drug utility in congestive heart failure due to the combination of decreased vascular resistance (α-adrenergic antagonism) and lack of reflex tachycardia (b-blockade). It should be noted that recent studies (15) involving low doses of selective b-blockers (e.g., metoprolol) display a similar benefit to carvedilol in reducing mortality in patients with heart failure.
Similar to carvedilol, labetalol (Figure 1), which contains four stereoisomers, is an adrenoceptor blocker with combined beta- and alpha-receptor blocking properties (9-11). However, the beta-blocking activity of the drug is between 3 to 7 folds greater than its alpha-blocking property (16). Additionally, different isomers contribute differently to the drug's a and b-blocking activities (9-11) (Table 1). Whereas the RR isomer is mostly responsible for the blocking activity of the drug, the SR isomer is most potent as a a-adrenoceptor blocker (9). Both the RS and SS isomers, on the other hand, show weak antagonistic activities against a and b receptors (9). Stereoselectivity in blocking beta adrenoceptors has also been reported (12) for nadolol (Figure 1), another beta-blocker with four stereoisomers (Table 1).
Stereoselectivity in Pharmacokinetics
Absorption
Generally, beta-blockers are absorbed from the gastrointestinal tract via passive diffusion. Therefore, their absorption is not considered stereoselective. However, some beta-blockers such as talinolol may undergo an intestinal secretion process that may be modestly stereoselective, resulting in an apparent nonlinearity in the kinetics of the drug with increasing oral doses (17). Nevertheless, despite the suggestion of an active intestinal secretion process, the overall pharmacokinetics of talinolol are not stereoselective (17).
Distribution
Beta-blockers are basic drugs that bind to both albumin and a1 -acid glycoprotein (AAG) in the plasma. Because of the asymmetric nature of proteins, binding of enantiomers to proteins resembles diastereomers. Therefore, the dissociation rate constant of binding may be stereoselective.
The information on the binding of some beta-blockers to plasma proteins is listed in Table 2. For acebutolol (18), pindolol (19), and sotalol (20), which have relatively high free fractions in plasma, the binding appears to be non-stereoselective (Table 2).
Table 2. Protein binding of individual enantiomers of beta blockers.
Drug
Study
Matrix
Percent Unbound
(:+)Unbound Ratio
(:+)Acebutolol
(18)
Human plasma (young)
86:84
1.0
Human plasma (elderly)
93:93
1.0
Pindolol
(19)
Human plasma
45:45
1.0
Propranolol
(21)
Human plasma
22.0:25.3
0.86
Human AAG
12.7:16.2
0.79
Human albumin
64.9:60.7
1.07
(22)
Human plasma
10.9:12.2
0.89
Human AAG
23.0:30.2
0.76
Human albumin
51.0:48.2
1.06
(23)
Human plasma (pregnant female)
20.7:22.4
0.92
Human plasma (fetus)
40.4:38.8
1.04
(24)
Human plasma (young)
11.7:18.0
0.65
Human plasma (elderly)
12.1:18.6
0.65
Human plasma (female)
10.9:17.8
0.61
Human plasma (male)
12.8:18.8
0.68
(25)
Human plasma (male)
9.1:10.8
0.84
Human plasma (female)
9.2:10.8
0.85
(26)
Human plasma
17.6:20.3
0.87
Sotalol
(20)
Human plasma (young)
96:96
1.0
Human plasma (elderly)
95:95
1.0
(47)
Human plasma (supraventricular tachyarrhythmia patients)
65:62
1.0
Abbreviation: AAG = a1-acid glycoprotein
However, stereoselective binding has been reported (21-26) for propranolol in both whole plasma as well as individual serum proteins (Table 2). As demonstrated in Table 2, the stereoselectivity in the binding of propranolol to human serum albumin is opposite of that observed for the human AAG. Whereas the free fraction of the (+)-enantiomer is higher in AAG, the opposite is true for albumin (Table 2). The overall stereoselectivity in the binding of propranolol to human serum, however, resembles that seen with AAG (Table 2).
A study of the propranolol binding in the maternal and fetal serum (23) further confirmed the importance of AAG in the overall extent of binding and stereoselectivity of propranolol (Table 2); a significantly lower concentration of AAG in the fetus blood (14 mg%), compared with that in the maternal blood (66 mg%), resulted in much higher free fractions and a change in the direction of stereoselectivity in binding in the fetal blood (Table 2).
The age and gender of the patients do not appear to have a substantial effect on the protein binding of propranolol enantiomers (Table 2). A modestly lower unbound fraction of (-)-propranolol in females (10.9%, Table 2) compared with males (12.8%, Table 2) reported in one study (24) was not observed in a subsequent study (25) which reported an unbound fraction of 9.1 and 9.2 in men and women, respectively (Table 2).
The tissue distribution of the enantiomers of propranolol (27-29) and pindolol (30) have been reported using laboratory animals. Collectively, these studies suggest that while the concentrations of the enantiomers of beta-blockers in different tissues may be stereoselective, the actual tissue uptake and binding of these drugs in most cases is non-stereoselective. The apparent stereoselectivity in the tissue concentrations of these drugs has been attributed mainly to the stereoselectivity in the plasma protein binding of the drugs. For example, Takahashi et al. (28) demonstrated that, compared with the (+)-enantiomer, the (-)-enantiomer of propranolol reaches higher concentrations in heart, muscle, gastrointestinal tract, kidney, brain, and lung of rats. However, this apparent stereoselectivity could be explained by higher free fraction of (-)-propranolol in plasma (28). Similarly, the stereoselective binding of the propranolol enantiomers to plasma proteins is the main reason behind an apparent stereoselectivity in the red blood cell distribution of the drug (31).
It has been shown that both hydrophilic (e.g. atenolol) and lipophilic (e.g. propranolol) beta-blockers are stored into and released from the adrenergic nerve endings (32). Further, it has been reported (32, 33) that the uptake into and release of atenolol from the models of adrenergic cells are stereoselective, favoring the more active (-)-enantiomer of atenolol by 2 to 5 fold. Additionally, a study in humans (34), chronically receiving racemic atenolol, indicated that the (-)-enantiomer of atenolol is selectively released into the plasma after exercise. The exercise-induced stereoselective release of atenolol from adrenergic nerve ending significantly changed the (+): (-) plasma concentration ratio of atenolol from 1.18 (at rest) to 0.64 (after exercise) (34). The effects of exercise on the stereoselective release of atenolol are apparently related to the duration of therapy because in disagreement with the above study using chronic dosing, the exercise-induced release of atenolol after a single dose of racemic atenolol was not stereoselective (35). In contrast to atenolol, the uptake of propranolol, a more lipophilic beta-blocker, into the adrenergic nerve cells or models appears to be via passive diffusion, and, therefore, is not stereoselective (33). Nonetheless, the stereoselectivity in the storage into and release of beta-blockers from the adrenergic nerve endings may have important clinical implications because it affects the concentration of the active enantiomer at its site of action.
Metabolism
The elimination of most beta-blockers occurs via hepatic metabolism and/or renal excretion of the unchanged drug. While the lipophilic beta-blockers, such as propranolol, are eliminated mostly by metabolism, the more hydrophilic beta-blockers, such as atenolol and nadolol, are almost exclusively excreted unchanged in urine. Some aspects of the stereospecific human metabolism of propranolol and metoprolol, two widely studied beta-blockers, will be discussed here.
In man, propranolol is metabolized by three main pathways of glucuronidation, ring hydroxylation, and side chain oxidation as depicted in Figure 2 (36). The ring hydroxylation process may occur at either position four or five (Fig. 2), both of them showing substrate stereoselectivity for R (+)-propranolol (36-38). The hydroxypropranolol is further conjugated with glucuronic acid, favoring the S (-)-enantiomer, or sulfate, favoring the R (+)-propranolol, before excretion into urine (32). As for N-dealkylation process (Fig. 2), the enantioselectivity in the metabolism appears to be related to the concentration of the drug; whereas at low substrate concentrations, the R (+)-enantiomer is preferentially metabolized, the opposite is true at high propranolol concentrations (36). Based on the urinary excretion of the propranolol glucuronides in humans, the formation of propranolol glucuronide, on the other hand, appears to favor S (-)-propranolol (37). Overall, the metabolism of propranolol is stereoselective for the less active R (+) enantiomer, resulting in a higher plasma concentrations of the S (-)-enantiomer in humans.
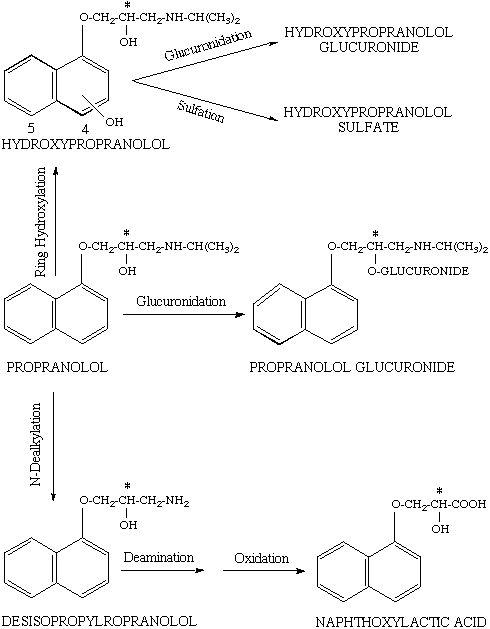
Figure 2. The schematic presentation of main metabolic pathways of propranolol in humans. Asterisk indicates the chiral center. Source: Ref. (36).
The metabolism of propranolol is affected by genetic polymorphism for both CYP1A (mephenytoin hydroxylation) and CYP2D6 (debrisoquine hydroxylation) isozymes in the liver (39, 40). Based on in vivo studies (39) in poor and extensive metabolizers of debrisoquine and mephenytoin and in vitro studies (40) using human liver microsomes and CYP isoforms, it appears that N-dealkylation of propranolol is mainly governed by S-mephenytoin-4-hydroxylase (CYP1A subfamily), whereas ring hydroxylation is predominantly related to debrisoquine isozyme (CYP2D6).
Metoprolol is another beta-blocker that is predominantly eliminated by hepatic metabolism (41). In humans, metoprolol is eliminated by several oxidation pathways, including benzylic hydroxylation a-hydroxylation) which results in an active metabolite and accounts for ~10% of the dose (42). This pathway is stereoselective for S (-)-metoprolol. The major metabolic pathway, however, is O-demethylation and further oxidation to a carboxylic acid metabolite that accounts for 65% of the dose (41). O-demethylation favors R (+)-metoprolol (42) and is responsible for the stereoselectivity observed in the plasma concentrations of metoprolol. A third metabolic pathway (N-dealkylation) accounts for < 10% of the dose in humans (42).
The oxidation of metoprolol cosegregates with debrisoquine hydroxylation, and debrisoquine phenotype significantly affects the stereoselective metabolism of the drug (41). The influence of debrisoquine hydroxylation phenotype on the pharmacokinetics and pharmacodynamics of both propranolol and metoprolol is described later in this review.
Renal excretion
As mentioned above, renal excretion of the unchanged drug is the major elimination pathway for hydrophilic beta-blockers such as atenolol, nadolol, and sotalol. In contrast to metabolism, the reported stereoselectivity in the renal clearance of beta-blockers is relatively low, with (-) :(+) renal clearance ratios being 0.90, 1.1, 1.2, and 1.05 for metoprolol (43), atenolol (44), pindolol (19) and sotalol (45), respectively. The small degree of stereoselectivity reported for the renal clearance of these drugs is most likely due to an active tubular secretion and/or reabsorption process. A stereoselective plasma protein binding, as a reason for the observed stereoselective renal clearance, is unlikely because the plasma protein bindings of pindolol and sotalol are not stereoselective (Table 2), and the protein binding of atenolol and nadolol enantiomers is negligible. Given the low degree of stereoselectivity in renal clearance, for those b-blockers subject to a large fraction of dose excreted in urine, a diminution of renal function might be expected to cause proportionately equal increases in plasma concentrations of both enantiomers.
Relative concentrations of enantiomers in plasma
The enantioselectivity in the plasma concentrations of beta-blockers is a reflection of all the processes (absorption, distribution, and elimination) involved in the pharmacokinetics of these drugs. The human kinetic parameters of various beta-blockers are listed in Tables 3-5. Except for metoprolol (Table 3), carvedilol (Table 3), and propranolol (Tables 4 and 5), the stereoselectivity in the pharmacokinetics of other studied beta-blockers is relatively modest (Table 3). For acebutolol, the active S (-)-enantiomer attains modestly higher plasma concentrations relative to its antipode (-:+ AUC ratio of 1.2 ± 0.1) (46). This stereoselectivity was suggested to be due to a stereoselective first pass metabolism in favor of the R (+)-enantiomer (46), as reflected in a slightly higher oral clearance of this enantiomer (Table 3).
Table 3. Stereospecific pharmacokinetics of some beta blockers in humans after single oral doses of the racemates.
Drug
(Age in yr, n)Dose, mg
Isomer
Cmax,
ng/mLTmax,
hAUC,
ng·h/mLCL,mL/min/kg
V,
L/kgCLR,
mL/mint1/2,
hrRef.
Acebutolol
(19-40, 12)200
()
246 ± 172
2.5 ± 0.9
1230 ±375
20 ± 5a
9.2 ± 2.6c
120 ± 38
5.4 ± 1.4
(46)
(+)
221 ± 155*
1030 ± 339*
25 ± 7a,*
11 ± 3.5c,*
124 ± 40
5.7 ± 1.8
Acebutolol
(60-75, 9)200
()
221 ± 106
2.4 ± 1.5
1380 ± 380
20 ± 5a
14 ± 8.9c
91 ± 36
7.6 ± 4.0
(18)
(+)
209 ± 91
1180 ± 359
24 ± 7a
14 ± 7.2c
90 ± 36
6.9 ± 3.3
Atenolol
(23-65, 6)50
()
226 ± 136
2.7 ± 1.1
1640 ± 602
1.5 ± 0.4b
0.88 ± 0.34
129 ± 32
6.13d
(44)
(+)
251 ± 138*
1860 ± 652*
1.4 ± 0.3b
0.79 ± 0.26
120 ± 29*
6.08d
Carvedilol
(53 ± 13, 13)25
()
34.2 ± 22.5
0.67e
125 ± 66
(69)
(+)
73.5 ± 44.3*
1.0e
288 ± 186*
Metoprolol,EM
(56 ± 1, 6)200
()
679 ± 388
70 ± 22
2.9 ± 1
(43)
(+)
408 ± 409*
75 ± 22*
2.8 ± 1
Metoprolol,PM
(58 ± 11, 6)200
()
3430 ± 623
56 ± 25
7.2 ± 1.5
(+)
3800 ± 635*
62 ± 26*
7.7 ± 1.7*
Pindolol
(19-36, 8)15
()
33 ± 7
1.7 ± 0.4
209 ± 73
222 ± 66
2.62d
(61)
(+)
36 ± 10
1.6 ± 0.5
244 ± 90*
170 ± 55*
2.85d
Sotalol
160
()
619 ± 164
3.1 ± 0.6
6760 ± 1200
158 ± 38
7.9 ± 1.2
(45)
(+)
615 ± 167
6950 ± 850
150 ± 25
8.2 ± 0.7
a Oral clearance or clearance/F
b Systemic clearance
c V/F
d Harmonic half life
e Median
* Significantly different from the ()-enantiomer.
Abbreviations: Cmax = maximum plasma concentration; Tmax = time to reach Cmax; AUC = area under the plasma concentration-time curve; CL= clearance; V= volume of distribution; CLR= renal clearance; t1/2 = plasma half life; EM, extensive metabolizers of debrisoquine; PM, poor metabolizers of debrisoquine; F = oral bioavailability.
Table 4. Stereospecific pharmacokinetics (mean ± SD) of propranolol in humans after intravenous doses of the racemate
Dose
Subjects
Isomer
CL, L/min
V, L
t1/2, hr
Reference
0.1 mg/kg
4 M, 1 F
21-38 yr()
1.03 ± 0.27
286 ± 52a,b
3.5 ± 0.5
(26)
(+)
1.21 ± 0.34*
337 ± 53a,b,*
3.6 ± 0.6
232 ± 28 µCi along with 16th dose of 80 mg t.i.d. po
12 M, White
()
0.77 ± 0.14
273 ± 32c
4.1 ± 0.5
(48)
(+)
0.84 ± 0.16
303 ± 45c
4.2 ± 0.3
13 M, Black
()
0.95 ± 0.27**
329 ± 98**
4.2 ± 0.8
(+)
1.1 ± 0.3**
397 ± 119**
4.3 ± 0.9
aVolume of distribution of the beta phase.
bBased on a 70-kg subject.
cSteady-state volume of distribution.
*Significantly different from the value for the ()-enantiomer.
**Significantly different from the value for the same isomer in the white group.
Abbreviations: CL= systemic clearance; V= volume of distribution; t1/2 = plasma half life.
Table 5. Stereospecific pharmacokinetics (mean ± SD) of propranolol in humans after single or multiple oral doses of the racemate (±) or pure enantiomers ( or +)
Drug, Dose (mg)
Subjects
Isomer
Cmax, ng/mL
AUC, ng·h/mL
CLO, L/min
t1/2, hr
Reference
(±), 80, single
5 M, 3 F, 24-27 yr
()
81.7 ± 31
329 ± 118
2.3 ± 0.7
4.5 ± 1.2
(53)
(+)
46.5 ± 25*
217 ± 114*
4.0 ± 2.0a,*
5.2 ± 2.4
(), 40, single
()
74.6 ± 16
274 ± 83**
2.7 ± 0.7a,**
4.4 ± 1.0
(±), 40, t.i.d.
10 M, 24 ± 1 yr
()
27 ± 18
119 ± 80
2.8
(55)
(+)
20 ± 15*
84 ± 67*
4.0
(), 20, t.i.d.
()
25 ± 14
98 ± 45
3.4
(±), 80, single
9 M, 28 ± 9, White
()
84.7 ± 37
523 ± 333
1.6 ± 0.8a
3.6 ± 0.9
(67)
(+)
61.9 ± 33
391 ± 296
2.5 ± 1.8a
3.6 ± 1
10 M, 27 ± 8, Chinese
()
49.8 ± 23***
351 ± 156***
2.6 ± 1.5a,***
4.0 ± 0.8
(+)
34.8 ± 26***
232 ± 111***
4.2 ± 2.5a,***
3.9 ± 0.8
(±), 80, t.i.d.
12 M, White
()
2.1 ± 0.5
4.1 ± 0.5
(48)
(+)
2.9 ± 0.9
4.2 ± 0.3
13 M, Black
()
3.3 ± 1.7***
4.2 ± 0.8
(+)
5.0 ± 4.2***
4.3 ± 0.9
(±), 80, t.i.d.
6 M, 6 F, 25-33 yr
()
387 ± 194
1.7
5.5 ± 2.4
(24)
(+)
329 ± 177*
2.0
4.3 ± 1.7
6 M, 6 F, 62-79 yr
()
475 ± 204
1.4
11.4 ± 5.9****
(+)
375 ± 187*
1.8
11.1 ± 4.8****
(±), 80, single
6 M, 24-32 yr
()
30 ± 11
152 ± 33
4.56 ± 0.9
3.6 ± 0.7
(63)
(+)
18 ± 6
100 ± 20
6.93± 1.5
4.3 ± 1.2
6 M, 65-80 yr
()
42 ± 19****
266 ± 118****
2.76 ± 1.2****
4.8 ± 0.5
(+)
27 ± 12****
171 ± 74****
4.55 ± 1.7****
4.8 ± 0.5
(±), 160, b.i.d.
15 M, 20-35 yr
()
290 ± 183
1600 ± 1040
1.5 ± 1.9a
3.3 ± 1.3
(54)
(+)
275 ± 183
1560 ± 1020
1.5 ± 1.8a
3.6 ± 1.2
(), 80, b.i.d.
()
267 ± 190
1590 ± 1410
1.3 ± 0.9a
3.0 ± 1.3
(+), 80, b.i.d.
(+)
212 ± 140
1120 ± 807
1.9 ± 1.8a
3.0 ± 1.2
aBased on a 70-kg subject.
*Significantly different from the value for the ()-enantiomer.
**Significantly different from the value for the ()-enantiomer after the administration of the racemate.
***Significantly different from the value for the same enantiomer in the white group.
****Significantly different from the value for the same enantiomer in the young group.
Abbreviations: Cmax = maximum plasma concentration; AUC = area under the plasma concentration-time curve; CLO= oral clearance (clearance/F); t1/2 = plasma half life; F = oral bioavailability
For atenolol and pindolol, two beta blockers with substantial elimination through renal excretion, a modest stereoselectivity in the renal clearance in favor of the S(-)-enantiomer results in slightly (< 20%) higher plasma concentrations of the less active R(+)-isomer (Table 3). Nevertheless, the stereoselectivity in the plasma concentrations of acebutolol, atenolol, and pindolol are perhaps of minor clinical significance.
After single doses of racemic sotalol to healthy volunteers, virtually no stereoselectivity was observed in the plasma concentrations of the enantiomers (45). The (-) :(+) ratios of maximum plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC) were 1.0 and 0.97, respectively, and the differences between enantiomers did not attain the level required for statistical significance (45). There was some stereoselectivity, however, noted in another study (47) when repeated doses of sotalol were administered to patients with supraventricular tachycardia. After at least 3 days of therapy with 80 or 160 mg of the racemate every 12 h, the (-):(+) ratios of steady-state AUC were 0.87 and 0.91 for the 80 and 160 mg doses, respectively. For both doses, the differences between enantiomers were statistically significant. It is not known why a greater degree of stereoselectivity appears to be present after repeated dose administration.
The stereoselectivity in the plasma concentrations of metoprolol is mostly related to the stereoselective metabolism and first pass effect of the drug. A slight stereoselectivity in the renal clearance of the drug in favor of R(+)-metoprolol (Table 3) virtually has no effect on the plasma stereoselectivity because of the negligible contribution of this pathway to the overall elimination of the drug. Whereas, a preferential metabolism of (+)-metoprolol in extensive metabolizers of debrisoquine results in a (-):(+) AUC ratio of 1.37 ± 0.32 (43), the stereoselectivity is reversed [(-) :(+) AUC ratio of 0.90 ± 0.06] in poor metabolizers of debrisoquine (Table 3).
For propranolol, a higher free fraction of the R (+)-enantiomer in blood (Table 2) results in a higher volume of distribution for this enantiomer after the IV administration of the racemate (Table 4). Additionally, the systemic clearance of R (+)-propranolol is slightly, but significantly, higher than that of S (-)-propranolol (Table 4), resulting in a slightly higher plasma concentration of S (-)-propranolol after the IV administration of the drug.
However, oral administration magnifies this stereoselectivity (Table 5), presumably due to a stereoselective first pass metabolism in favor of R (+)-propranolol (48). After the oral administration of the racemate, the (-) :(+) AUC ratios for propranolol ranged from 1.0 to 1.6 in various studies (Table 5). The possible reasons for this wide range of ratios are explained in the following section.
Factors Affecting the Pharmacokinetics and Pharmacodynamics of Beta Blockers
Various factors related to the drug and the patient may affect the stereospecific pharmacokinetics and pharmacodynamics of beta-blockers. Factors related to the drug include the dosing rate of the drug and the interactions between the enantiomers themselves and between the enantiomers and other drugs. Factors related to the patient may be the age and gender, racial background, disease states, and the patient metabolic phenotype.
Input rate
Theoretically (49, 50), the oral rate of input of racemic drugs with stereoselective metabolism may affect the plasma concentration ratio of the enantiomers. This is because the rate of input of the drug into the portal vein may have a different effect on the degree of saturation of the metabolic pathways of the enantiomers, with resultant stereoselectivity in the first pass metabolism of these drugs. Indeed, as early as 1982, Silber et al. (51) reported that the (-):(+) steady state plasma concentration ratios of propranolol significantly decreased with an increase in the daily dose of the drug; the ratios (mean ± SD) were 2.45 ± 1.12, 1.78 ± 0.60, and 1.51 ± 0.05 after the oral multiple doses of 160, 240, and 320 mg/day, respectively. The same trend is observed when results of several studies are combined in Figure 3. The input rate-dependent change in the ratio may be attributed to a dose-dependent saturation of the first pass metabolism of propranolol with a greater saturation for the R (+)-enantiomer.
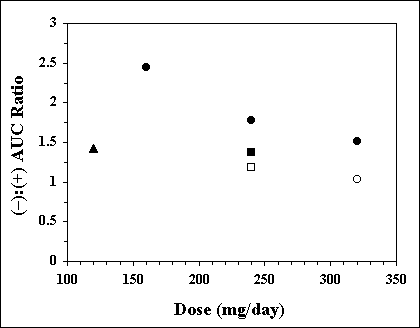
Figure 3. The average steady state (-):(+) AUC ratio of propranolol as a function of daily dose of the racemic drug in different studies. Keys: l, Ref. (51); p, Ref. (55); n, Ref. (67); o, Ref. (24); m, Ref. (54)
In contrast to Silber et al. (51), Bleske et al. (52) could not detect any significant effect of input rate on the stereoselectivity in the propranolol pharmacokinetics. The lack of effect of input rate in the latter study is perhaps because only single and relatively low doses of the drug were used (52). Simulations (49, 50) have shown that the effects of input rate on the stereoselective pharmacokinetics are significant in the non-linear input ranges when the input rate approaches the maximum velocity of the metabolism. Additional pharmacokinetic/dynamic studies are needed to determine the clinical significance of the effects of input rate on the stereoselective metabolism of propranolol.
Interactions
The two enantiomers of a racemic drug may interact with each other at different pharmacokinetic or pharmacodynamic levels. This type of interaction has been studied for atenolol (3) and propranolol (53-55). For atenolol, there was no pharmacokinetic or pharmacodynamic interaction between the two enantiomers; the half-dosed S (-)-atenolol produced the same effect as did the racemic atenolol (3). Additionally, the plasma concentration-time profiles of S (-)-atenolol were identical after the administration of the racemate or the half-dosed pure enantiomer. On the other hand, both single (53) and multiple (54) dose studies have shown that there is a significant interaction between the enantiomers of propranolol. When administered as pure enantiomer, as opposed to the racemate, R (+)-propranolol tends to show lower plasma concentrations (54). However, the kinetics of the more active S (-)-enantiomer appear to be the same whether it is administered as a pure enantiomer or racemate (53-55).
In addition to enantiomer-enantiomer interactions, a racemic drug may interact with other drugs stereoselectively. For instance, stereoselective interactions have been reported in man between propranolol and calcium channel blockers (56, 57), cimetidine (58), and quinidine (36). Calcium channel blockers nicardipine (56), diltiazem (57), and verapamil (57) all decreased the first-pass metabolism of both enantiomers of propranolol. However, this inhibitory effect was stereoselective for the R (+)-enantiomer in the case of both verapamil (57) and nicardipine (56), resulting in a significant increase in the (+) :(-) AUC ratios in plasma. In terms of effects, nicardipine did not increase the blood pressure reduction effect of propranolol (56), a phenomenon that may be explained by a more significant pharmacokinetic effect on the less active R (+) enantiomer. Similarly, cimetidine decreased the oral clearance of R (+)-propranolol to a more significant degree than that of the S (-)-enantiomer (58). As for quinidine, human liver microsome studies (36) indicated that this selective inhibitor of CYPD26 reduced the ring hydroxylation of propranolol in a stereoselective manner in favor of R (+)-propranolol. This was in agreement with in vivo studies (59) showing 180% and 100% increases in the plasma AUCs of R (+) - and S (-)-propranolol, respectively, because of quinidine co-administration. Interestingly, all these studies have shown that the inhibition of the metabolism of propranolol by different drugs is stereoselective for R (+)-propranolol.
In addition to the inhibition of the metabolism of propranolol, verapamil reportedly (60) inhibits the metabolism of metoprolol, another extensively metabolized beta-blocker. As mentioned in the metabolism section, the O-demethylation pathway is the main metabolic pathway for the metabolism of metoprolol accounting for 65% of the dose. Verapamil significantly inhibits this pathway in a stereoselective manner favoring inhibition of the metabolism of R (+)-metoprolol (60). The interaction of verapamil and metoprolol results in clinically significant adverse reactions, presumably due to higher plasma concentrations of metoprolol.
Cimetidine not only reduces the metabolism of beta-blockers such as propranolol, it is also known to act as an inhibitor of tubular secretion of a number of organic cations. Therefore, it is not surprising to see that the renal clearance of pindolol is substantially and stereoselectively reduced by co-administration of this drug (61). The administration of 400 mg cimetidine twice a day for 2 days before and 2 days after pindolol administration resulted in 26% and 34% reductions in the renal clearances of S(-)- and R(+)-pindolol, respectively. Therefore, the plasma concentrations of the R (+)-enantiomer increased more drastically (47%) than those of the S (-)-enantiomer (38%) in the presence of cimetidine (61). Because renal clearance accounts for only 50% of pindolol elimination, the significant increases in the plasma concentrations of pindolol enantiomers because of cimetidine co-administration cannot be explained based on the inhibition of its renal clearance only. Apparently, cimetidine also reduces the metabolism of pindolol.
Age and gender
The effect of age on the stereospecific pharmacokinetics of propranolol has been the subject of several studies (24, 62-64) with conflicting results. For instance, while Colangelo et al. (64) and Lalonde et al. (62) reported no significant changes in the oral clearances of the propranolol enantiomers with advancing age, others (24, 63) have reported a significant decline in the clearance of both enantiomers in the elderly. Additionally, in contrast to Zhou et al. (63) who showed no significant differences in the plasma half lives of the propranolol enantiomers between the young and the elderly, Gilmore et al. (24) reported that the half lives of both propranolol enantiomers in the elderly were more than two-fold longer than those in the young. These conflicting results are mostly due to different designs of these studies with regard to the dose, duration of therapy (single versus multiple dosing), duration of sampling, and the number of subjects used. Overall, these studies suggest that the clearance of both enantiomers of propranolol is reduced in the elderly. Additionally, in the presence of extended duration of sampling (e.g. =24 hr), the terminal half lives of the propranolol enantiomers are apparently longer in the elderly than in the young. However, the reduction in the oral clearance and prolongation of plasma half-life appear to be to the same extent for both enantiomers. These data indicate that the known decreased response to beta-blockers in the elderly subjects cannot be explained by stereoselective pharmacokinetic differences between the young and elderly.
For acebutolol, an advance in the age resulted in a de-crease in creatinine clearance and an associated decrease in the renal clearance of both enantiomers of the parent drug and its metabolite diacetolol (18). However, the decrease in the renal clearance of the enantiomers was not stereoselective. On the other hand, a reduction in creatinine clearance with advancing age was associated with a significant decrease in the (-):(+) AUC ratio of the drug (from 1.3 to 1.1 when creatinine clearance declined from 90 to 45 mL/min), suggesting that aging has a stereoselective effect on the other pathways (e.g. metabolism) of acebutolol elimination.
In terms of gender, non-stereoselective studies (65) have clearly shown that the oral clearance, and not the systemic clearance, of propranolol is significantly (63%) higher in men than in women. This difference was attributed to significant increases in the side-chain oxidation and glucuronidation of propranolol during the first pass metabolism in men (65). Available stereoselective studies (24, 25), however, have failed to show a significant difference between males and females in the main pharmacokinetic parameters of propranolol. Again, this discrepancy may be related to methodological differences and the power of statistical tests used in these studies. Future, well-designed studies are needed to test the effects of gender on the stereospecific pharmacokinetics of propranolol.
The importance of delineation of the stereospecific pharmacokinetics in explaining gender-related pharmacodynamics was demonstrated recently for labetalol (66). Labetalol dose was titrated to a specific antihypertensive effect in 14 men and 5 women with ages ranging from 40 to 63 and 40 to 56 years, respectively. The dose-corrected AUC values for total labetalol in women were 80% higher than those in men. However, the antihypertensive effects were the same for both groups. This discrepancy could be easily explained by stereoselective differences in the pharmacokinetics of labetalol isomers in men and women; whereas the concentrations of the alpha blocking isomer (SR) and two relatively inactive isomers of labetalol (SS and RS) were between 60 to 80% higher in women, the plasma concentrations of the main beta-blocking isomer (RR) were the same in both groups, resulting in similar antihypertensive effects in men and women.
Genetic factors
Ethnic background: It is known that Chinese subjects are more responsive than white subjects to the same dose or plasma concentrations of racemic propranolol. A study (67) investigated whether this could be attributed to higher (-) :(+) AUC ratios in Chinese subject, compared with the ratio in white population. Although the plasma concentrations of both enantiomers were substantially lower in Chinese volunteers, the ratio was the same in both populations (67), suggesting a pharmacodynamic difference in these two populations with regard to the beta-blockade effect.
In contrast to the Chinese, the black population responds less to the same dose of propranolol, when compared with the white population. Sowinski et al. (48) showed that both the systemic and oral clearances of both enantiomers of propranolol are substantially higher in blacks than whites. This difference was mostly attributed to a higher intrinsic clearance of propranolol enantiomers, in association with a slightly lower (9%) hepatic blood flow in blacks. The limited available information on the effects of ethnicity on the pharmacokinetics of propranolol suggests that the racial differences in the effects of this drug cannot be attributed to the stereoselectivity in the pharmacokinetics of the drug. Rather, these differences may be due to pharmacodynamic differences among ethnic populations.
Metabolic phenotype of the patient: As mentioned in the Metabolism section, the metabolic phenotype of patients may significantly affect the metabolism and, consequently, the overall pharmacokinetics and pharmacodynamics of propranolol and metoprolol. For propranolol, Ward et al. (39) reported that although the ring hydroxylation and N-dealkylation processes were deficient in poor metabolizers of debrisoquine and mephenytoin, respectively, the overall oral clearance of the propranolol enantiomers were modestly reduced in volunteers with one of these deficiencies (Table 6). Additionally, the stereoselectivity in the plasma concentrations in these volunteers were not different than that in extensive metabolizers (Table 6). However, a combined poor metabolism of mephenytoin and debrisoquine (observed only in one volunteer) substantially reduced the oral clearance of both enantiomers and abolished the stereoselectivity in the plasma concentration of propranolol (Table 6). It should be noted that the cosegregation of both deficiencies is expected to be very low (0.4% of the population) (39).
Table 6. Oral clearance and plasma half lives of propranolol enantiomers after oral administration of the racemate (80 mg) to volunteers with different phenotypes of debrisoquine and mephenytoin hydroxylation.a
Group
n
Oral Clearance, ml/min
(+)-Propranolol
()-Propranolol
(+):() Ratio
EM
6
2670 ± 697
1910 ± 632
1.42 ± 0.53
PMD
4
1860 ± 1110
1420 ± 788
1.25 ± 0.06
PMM
5
2010 ± 909
1401 ± 595
1.42 ± 0.09
PMD/M
1
918
850
1.08
aSource: Reference (39).
Abbreviations: EM, extensive metabolizers of debrisoquine and mephenytoin; PMD, poor metabolizers of debrisoquine and extensive metabolizers of mephenytoin; PMM, poor metabolizers of mephenytoin and extensive metabolizers of debrisoquine; PMD/M, poor metabolizers of both debrisoquine and mephenytoin.
For metoprolol, Lennard et al. (41) demonstrated that after a 200-mg oral dose, the plasma concentrations of racemic metoprolol in poor metabolizers of debrisoquine were 6 times higher than those in the extensive metabolizers. Additionally, when the beta blockade effect was plotted against the plasma concentrations of the racemic drug, the relationship was shifted to the right in poor metabolizers (43). This discrepancy was attributed to the differences in the stereoselectivity in the metabolism and plasma concentrations of the drug in the two groups. Whereas the (-) :(+) metoprolol AUC ratio was 1.37 ± 0.32 (mean ± SD) in the extensive metabolizers, the ratio was 0.90 ± 0.06 in poor metabolizers (43). Therefore, the same concentrations of the racemate would contain less of the active (-)-enantiomer in poor metabolizers, shifting the effect-concentration relationship to the right.
Although the metabolism of both propranolol and metoprolol are affected by debrisoquine phenotype, the above data clearly shows that the effect is more substantial for metoprolol. Debrisoquine polymorphism also significantly affects the kinetics of another beta-blocker, bufuralol (68). In contrast to metoprolol, however, poor metabolism of debrisoquine intensified the stereoselectivity in the plasma concentrations of bufuralol [(-):(+) plasma concentration ratio of 1.8 and 2.6 at 3 hr in extensive and poor metabolizers, respectively] (68). This was due to a significant reduction of the metabolism of (-)-bufuralol by ring hydroxylation in poor metabolizers of debrisoquine (68). Therefore, the effect of debrisoquine hydroxylation phenotype on the degree and direction of stereoselectivity in pharmacokinetics varies with each beta-blocker.
Congestive heart failure
For highly metabolized drugs, altered cardiac output may influence the drug plasma concentrations in the presence of moderate to high hepatic extraction ratio, conditions that are true for carvedilol enantiomers (69). Tenero et al. (13) demonstrated that in patients with Class III or IV congestive heart failure, the plasma concentrations of carvedilol enantiomers were substantially higher than those in healthy volunteers. Additionally, patients with Class IV congestive heart failure had consistently higher plasma concentrations (up to two-fold) than those patients with Class III congestive heart failure (13). Similar to healthy volunteers, patients with congestive heart failure exhibited stereoselectivity in both Cmax and AUC of the drug (13). However, the R:S AUC ratios observed in these patients (~1.9) were lower than those observed in otherwise healthy young (2.8) and elderly (2.3) volunteers (13), suggesting that the effects of congestive heart failure on the pharmacokinetics of carvedilol are stereoselective.
Although it was not mentioned by the authors, there does appear to be a difference in the level of stereoselectivity in AUC between the patients with Class III and Class IV congestive heart failure (13). The reported mean R(+):mean S(-) AUC in the Class IV patients were consistently higher (20-43%) than the corresponding ratios in the patients with Class III congestive heart failure across a dosage range of 6.25-50 mg administered every 12 hr. The ratios of mean R(+):mean S(-) Cmax were also consistently higher in the Class IV patients (14-18%). Overall, these data suggest that Class IV congestive heart failure patients attain higher plasma concentrations of both enantiomers, in favor of the R-enantiomer, when compared with Class III patients.
Conclusion
For the most part, the enantiomers of the b-adrenergic antagonists share similarities with respect to pharmacologic effects, with the (-)-enantiomer usually possessing a substantially higher ability to bind to b-adrenergic receptors. In some cases, such as sotalol, carvedilol, and labetalol, the enantiomers may possess other qualities that add to the beta-blocking properties of the drug. As for pharmacokinetics, there does not seem to be a general pattern in the degree and/or direction of stereoselectivity among different beta-blockers. This is perhaps because b-adrenergic antagonists encompass a wide spectrum of pharmacokinetic properties, with low to high degrees of plasma protein binding, extent of excretion into urine as unchanged drug, and hepatic extraction ratios. Additionally, the degree and direction of stereoselectivity in the pharmacokinetics of these drugs are susceptible to change because of patient and/or disease characteristics. Because of the significant enantioselectivity in the pharmacologic effects of beta-blockers, a stereoselective change in the pharmacokinetics of these drugs may be associated with an altered pharmacodynamic profile. An understanding of the stereospecific pharmacokinetics and pharmacodynamics of beta-blockers may help clinicians to interpret and predict differences among patients in pharmacologic response to these drugs.
References
Benet LZ; Oie S; Schwartz JB. Design and optimization of dosage regimens; pharmacokinetic data, in Hardman JG: Limbird LE: Molinoff PB: Rudden RW: Goodman Gilman A (eds.), Goodman & Gilman's The Pharmacological Basis of Therapeutics. McGraw-Hill, New York, pp 1707-1792, 1996.
Pearson AA; Gaffney TE; Walle T; Privitera PJ. A stereoselective central hypotensive action of atenolol. J Pharmacol Exp Ther, 250:759-763, 1989.
Stoschitzky K; Egginger G; Zernig G; Klein W; Lindner W. Stereoselective features of (R)- and (S)- atenolol: clinical pharmacological, pharmacokinetic, and radioligand binding studies. Chirality, 5:15-19, 1993.
Nathanson JA. Stereospecificity of beta adrenergic antagonists: R-enantiomers show increased selectivity for beta-2 receptors in ciliary process. J Pharmacol Exp Ther, 245:94-101, 1988.
Weyl JD; Snyder RW; Hanson RC. Differential cardioprotective properties of the l- and d- enantiomers of bucindolol in a canine model of heart failure. Arch Int Pharmacodyn Ther, 275:4-12., 1985.
Jeppsson AB; Johansson U; Waldeck B. Steric aspects of agonism and antagonism at beta-adrenoceptors: experiments with the enantiomers of terbutaline and pindolol. Acta Pharmacol Toxicol, 54:285-91., 1984.
Barrett A; Cullum V. The biological properties of the optical isomers of propranolol and their effects on cardiac arrhythmias. Br J Pharmacol, 34:43-55, 1968.
Kato R; Ikeda N; Yabek SM; Kannan R; Singh BN. Electrophysiologic effects of the levo- and dextrorotatory isomers of sotalol in isolated cardiac muscle and their in vivo pharmacokinetics. J Am Coll Cardiol, 7:116-25., 1986.
Brittain RT; Drew GM; Levy GP. The alpha- and beta-adrenoceptor blocking potencies of labetalol and its individual stereoisomers in anaesthetized dogs and in isolated tissues. Br J Pharmacol, 77:105-14, 1982.
Gold EH; Chang W; Cohen M; Baum T; Ehrreich S; Johnson G; Prioli N; Sybertz EJ. Synthesis and comparison of some cardiovascular properties of the stereoisomers of labetalol. J Med Chem, 25:1363-70, 1982.
Riva E; Mennini T; Latini R. The alpha- and beta-adrenoceptor blocking activities of labetalol and its RR-SR (50:50) stereoisomers. Br J Pharmacol, 104:823-8, 1991.
Srinivas NR; Barr WH; Shyu WC; Mohandoss E; Chow S; Staggers J; Balan G; Belas FJ; Blair IA; Barbhaiya RH. Bioequivalence of two tablet formulations of nadolol using single and multiple dose data: assessment using stereospecific and nonstereospecific assays. J Pharm Sci, 85:299-303, 1996.
Tenero D; Boike S; Boyle D; Ilson B; Fesniak HF; Brozena S; Jorkasky D. Steady-state pharmacokinetics of carvedilol and its enantiomers in patients with congestive heart failure. J Clin Pharmacol, 40:844-853, 2000.
Bartsch W; Sponer G; Strein K; Muller-Beckmann B; Kling L; Bohm E; Martin U; Borbe HO. Pharmacological characteristics of the stereoisomers of carvedilol. Eur J Clin Pharmacol, 38:S104-7., 1990.
Smith AJ; Wehner JS; Manley HJ; Richardson AD; Beal J; Bryant PJ. Current role of beta-adrenergic blockers in the treatment of chronic congestive heart failure. Am J Health Syst Pharm, 58:140-5., 2001.
Lalonde RL; O'Rear TL; Wainer IW; Drda KD; Herring VL; Bottorff MB. Labetalol pharmacokinetics and pharmacodynamics: evidence of stereoselective disposition. Clin Pharmacol Ther, 48:509-519, 1990.
Wetterich U; Spahn-Langguth H; Mutschler E; Terhaag B; Rosch W; Langguth P. Evidence for intestinal secretion as an additional clearance pathway of talinolol enantiomers: concentration- and dose-dependent absorption in vitro and in vivo. Pharm Res, 13:514-522, 1996.
Piquette-Miller M; Foster RT; Kappagoda CT; Jamali F. Effect of aging on the pharmacokinetics of acebutolol enantiomers. J Clin Pharmacol, 32:148-156, 1992.
Hsyu P-H; Giacomini KM. Stereoselective renal clearance of pindolol in humans. J Clin Invest, 76:1720-1726, 1985.
Carr RA; Pasutto FM; Lewanczuk RZ; Foster RT. Protein binding of sotalol enantiomers in young and elderly human and rat serum using ultrafiltration. Biopharm Drug Dispos, 16:705-712, 1995.
Walle U; Walle T; Bai S; Olanoff L. Stereoselective binding of propranolol to human plasma, α1-acid glycoprotein, and albumin. Clin Pharmacol Ther, 34:718-723, 1983.
Albani F; Riva R; Contin M; Baruzzi A. Stereoselective binding of propranolol enantiomers to human α1-acid glycoprotein and human plasma. Br J Clin Pharmacol, 18:244-246, 1984.
Belpaire FM; Wynant P; Vantrappen P; Dhont M; Verstraete A; Bogaert MG. Protein binding of propranolol and verapamil enantiomers in maternal and foetal serum. Br J Clin Pharmacol, 39:190-193, 1995.
Gilmore D; Gal J; Gerber J; Nies A. Age and gender influence the stereoselective pharmacokinetics of propranolol. J Pharmacol Exp Ther, 261:1181-1186, 1992.
Walle U; Fagan T; Topmiller M; Conradi E; Walle T. The influence of gender and sex steroid hormones on the plasma binding of propranolol enantiomers. Br J Clin Pharmacol, 37:21-25, 1994.
Olanoff L; Walle T; Walle K; Cowart T; Gaffney T. Stereoselective clearance and distribution of intravenous propranolol. Clin Pharmacol Ther, 35:755-761, 1984.
Walle UK; Thibodeaux H; Privitera PJ; Walle T. Stereochemistry of tissue distribution of racemic propranolol in the dog. Chirality, 1:192-6, 1989.
Takahashi H; Ogata H; Kanno S; Takeuchi H. Plasma protein binding of propranolol enantiomers as a major determinant of their stereoselective tissue distribution in rats. J Pharmacol Exp Ther, 252:272-278, 1990.
Tamai G; Edani M; Imai H. Chiral separation and determination of propranolol enantiomers in rat or mouse blood and tissue by column switching high performance liquid chromatography with ovomucoid bonded stationary phase. Biomed Chromatogr, 4:157-160, 1990.
Yan H; Lewander T. Differential tissue distribution of the enantiomers of racemic pindolol in the rat. Eur Neuropsychopharmacol, 10:59-62, 1999.
Takahashi H; Ogata H. Plasma protein binding and blood cell distribution of propranolol enantiomers in rats. Biochem Pharmacol, 39:1495-1498, 1990.
Walle T; Webb JG; Bagwell EE; Walle UK; Daniell HB; Gaffney TE. Stereoselective delivery and actions of beta receptor antagonists. Biochem Pharmacol, 37:115-124, 1988.
Walle T; Webb JG; Walle UK; Bagwell EE. Stereoselective accumulation of the β-receptor blocking drug atenolol by human platelets. Chirality, 3:451-453, 1991.
Stoschitzky K; Lindner W; Klein W. Stereoselective release of (S)-atenolol from adrenergic nerve endings at exercise. Lancet, 340:696-697, 1992.
Stoschitzky K; Kahr S; Donnerer J; Schumacher M; Luha O; Maier R; Klein W; Lindner W. Stereoselective increase of plasma concentrations of the enantiomers of propranolol and atenolol during exercise. Clin Pharmacol Ther, 57:543-551, 1995.
Marathe PH; Shen DD; Nelson WL. Metabolic kinetics of pseudoracemic propranolol in human liver Microsomes. Enantioselectivity and quinidine inhibition. Drug Metab Dispos, 22:237-247, 1994.
Walle T. Stereoselectivity of the in vivo disposition and metabolism of propranolol in dog and man using deuterium-labeled pseudoracemates. Drug Metab Dispos, 13:279-282, 1985.
Nelson W; Shetty H. Stereoselective oxidative metabolism of propranolol in the microsomal fraction from rat and human liver. Use of deuterium labeling and pseudoracemic mixtures. Drug Metab Dispos, 14:506-508, 1986.
Ward SA; Walle T; Walle UK; Wilkinson GR; Branch RA. Propranolol's metabolism is determined by both mephenytoin and debrisoquin hydroxylase activities. Clin Pharmacol Ther, 45:72-79, 1989.
Yoshimoto K; Echizen H; Chiba K; Tani M; Ishizaki T. Identification of human CYP isoforms involved in the metabolism of propranolol enantiomers - N-desisopropylation is mediated mainly by CYP1A2. Br J Clin Pharmacol, 39:421-431, 1995.
Lennard MS; Silas JH; Freestone S; Ramsay LE; Tucker GT; Woods HF. Oxidation phenotype--a major determinant of metoprolol metabolism and response. N Engl J Med, 307:1558-60, 1982.
Murthy SS; Shetty U; Nelson WL; Jackson PR; Sennard MS. Enantioselective and diastereoselective aspects of the oxidative metabolism of metoprolol. Biochem Pharmacol, 40:1637-1644, 1990.
Lennard MS; Tucker GT; Silas JH; Freestone S; Ramsay LE; Woods HF. Differential stereoselective metabolism of metoprolol in extensive and poor debrisoquine metabolizers. Clin Pharmacol Ther, 34:732-737, 1983.
Mehvar R; Gross ME; Kreamer RN. Pharmacokinetics of atenolol enantiomers in humans and rats. J Pharm Sci, 79:881-885, 1990.
Carr RA; Foster RT; Lewanczuk RZ; Hamilton PG. Pharmacokinetics of sotalol enantiomers in humans. J Clin Pharmacol, 32:1105-1109, 1992.
Piquette-Miller M; Foster RT; Kappagoda CT; Jamali F. Pharmacokinetics of acebutolol enantiomers in humans. J Pharm Sci, 80:313-316, 1991.
Fiset C; Philippon F; Gilbert M; Turgeon J. Stereoselective disposition of (±)- sotalol at steady-state conditions. Br J Clin Pharmacol, 36:75-77, 1993.
Sowinski KM; Lima JJ; Burlew BS; Massie JD; Johnson JA. Racial differences in propranolol enantiomer kinetics following simultaneous iv and oral administration. Br J Clin Pharmacol, 42:339-346, 1996.
Mehvar R. Input rate-dependent stereoselective pharmacokinetics: effect of pulsatile oral input. Chirality, 6:185-195, 1994.
Mehvar R. Input rate-dependent stereoselective pharmacokinetics: enantiomeric oral bioavailability and blood concentration ratios after constant oral input. Biopharm Drug Dispos, 13:597-615, 1992.
Silber B; Holford NHG; Riegelman S. Stereoselective disposition and glucuronidation of propranolol in humans. J Pharm Sci, 71:699-704, 1982.
Bleske BE; Welage LS; Rose S; Amidon GL; Shea MJ. The effect of dosage release formulations on the pharmacokinetics of propranolol stereoisomers in humans. J Clin Pharmacol, 35:374-378, 1995.
Lindner W; Rath M; Stoschitzky K; Semmelrock H. Pharmacokinetic data of propranolol enantiomers in a comparative human study with (S)- and (R,S)-propranolol. Chirality, 1:10-13, 1989.
Paradiso-Hardy FL; Walker SE; Bowles SK. Steady-state pharmacokinetics of propranolol enantiomers in healthy male volunteers. Int J Clin Pharm Therapeutics, 36:370-375, 1998.
Stoschitzky K; Linder W; Egginger G; Brunner F; Obermayer-Pietsch; Passath A; Klein W. Racemic (R,S)-propranolol versus half-dosed optically pure (S)-propranolol in humans at steady state: Hemodynamic effects, plasma concentrations, and influence on thyroid hormone levels. Clin Pharmacol Ther, 51:445-453, 1992.
Vercruysse I; Belpaire FM; Wynant P; Massart DL; Dupont AG. Enantioselective inhibitory effect of nicardipine on the hepatic clearance of propranolol in man. Chirality, 6:5-10, 1994.
Hunt BA; Bottorff MB; Herring VL; Self TH; Lalonde RL. Effects of calcium channel blockers on the pharmacokinetics of propranolol stereoisomers. Clin Pharmacol Ther, 47:584-91, 1990.
Donn K; Powell J; Wainer I. Stereoselectivity of cimetidine inhibition of propranolol oral clearance. Clin Pharmacol Ther, 37:191, 1985.
Zhou H-H; Anthony LB; Roden DM; Wood AJ. Quinidine reduces clearance of (+)-propranolol more than (-)-propranolol through marked reduction in 4-hydroxylation. Clin Pharmacol Ther, 47:686-693, 1990.
Kim M; Shen D; Eddy A; Nelson W. Inhibition of the enantioselective oxidative metabolism of metoprolol by verapamil in human liver microsomes. Drug Metab Dispos, 21:309-317, 1993.
Somogyi AA; Bochner F; Sallustio BC. Stereoselective inhibition of pindolol renal clearance by cimetidine in humans. Clin Pharmacol Ther, 51:379-387, 1992.
Lalonde RL; Tenero DM; Burlew BS; Herring VL; Bottorff MB. Effects of age on the protein binding and disposition of propranolol stereoisomers. Clin Pharmacol Ther, 47:447-55, 1990.
Zhou H-H; Whelan E; Wood JJ. Lack of effect of ageing on the stereochemical disposition of propranolol. Br J Clin Pharmacol, 33:121-123, 1992.
Colangelo P; Blouin R; Steinmetz J; McNamara P; DeMaria A; Wedlund P. Age and propranolol stereoselective disposition in humans. Clin Pharmacol Ther, 51:489-494, 1992.
Walle T; Walle U; Cowart T; Conradi E. Pathway-selective sex differences in the metabolic clearance of propranolol in human subjects. Clin Pharmacol Ther, 46:257-263, 1989.
Johnson JA; Akers WS; Herring VL; Wolfe MS; Sullivan JM. Gender differences in labetalol kinetics: importance of determining stereoisomer kinetics for racemic drugs. Pharmacotherapy, 20:622-8, 2000.
Zhou H-H; Wood AJ. Differences in stereoselective disposition of propranolol do not explain sensitivity differences between white and Chinese subjects: correlation between the clearance of (-)- and (+)-propranolol. Clin Pharmacol Ther, 47:719-723, 1990.
Dayer P; Leemann T; Kupfer A; Kronbach T; Meyer UA. Stereo- and regioselectivity of hepatic oxidation in man - effect of the debrisoquine/sparteine phenotype on bufuralol hydroxylation. Eur J Clin Pharmacol, 31:313-318, 1986.
Gehr TW; Tenero DM; Boyle DA; Qian Y; Sica DA; Shusterman NH. The pharmacokinetics of carvedilol and its metabolites after single and multiple dose oral administration in patients with hypertension and renal insufficiency. Eur J Clin Pharmacol, 55:269-77., 1999.
Corresponding Author: Reza Mehvar, Ph.D., School of Pharmacy, Texas Tech University Health Sciences Center, 1300 Coulter, Amarillo, TX 79106. rmehvar@ama.ttuhsc.edu
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps
