J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 4(1):32-41, 2001
Molecular Modeling of Arginine-Glycine-Aspartic Acid (RGD) Analogs: Relevance to Transepithelial Transport
Rudraksh Sharan
Department of Pharmaceutics, and Thad Cochran Center for the Development of Natural Products, University of Mississippi, University, Mississippi, USAWerner Rubas
COR Therapeutics, Inc., South San Francisco, California, USAWilliam M. Kolling
College of Pharmacy, Department of Basic Pharmaceutical Sciences, University of Louisiana at Monroe, Monroe, Louisiana, USAHamidreza Ghandehari1
Department of Pharmaceutical Sciences, University of Maryland School of Pharmacy, Baltimore, Maryland, USAReceived February 15, 2001, Revised March 29, 2001, Accepted March 30, 2001
ABSTRACT
Purpose . The aim of this research is to model the effect of methylation on hydrogen bonding ability, surface area, polar surface area, volume, lipophilicity, charge, and cross-sectional diameters of a series of mono-, di-, and tri- methyl substituted analogs of arginine-glycine-aspartic acid (RGD) and compare these parameters to in vitro transport properties across Caco-2 monolayers.
Methods . Molecular modeling was used to investigate the structural parameters that may influence the transport properties of RGD and its methyl analogs at pH 7.4. Log P was experimentally determined using a potentiometric method and compared to cLogP. Transport studies were carried out using Caco-2 cell monolayers.
Results . Parameters such as polar and total surface area, volume, and Log P were found to vary with both the number and the sites of methyl substitution on the RGD molecule. The calculated as well as the experimental Log P values were found to be less than minus 2. The calculated maximum cross-sectional diameters ranged from 9 to 12 Å. No detectable transport was noted.
Conclusions . Results of our study indicate that in the design considerations for the development of new peptidomimetic RGD analogs with enhanced oral bioavailability, an important parameter to consider is the three dimensional conformation of the peptides which influences their hydrogen bonding ability, polarity and molecular geometry.
INTRODUCTION
Recent research has focussed on the development of arginine-glycine-aspartic acid (RGD) containing anti-thrombotic agents that interfere with platelet aggregation at a final common pathway, namely the platelet glycoprotein GPIIb/IIIa complex (1). An unresolved issue, particularly important for chronic anti-thrombotic therapy, is the lack of adequate oral bioavailability of the recently developed peptide GPIIb/IIIa antagonists (2). Among the factors that impede the oral bioavailability of peptide drugs are the enzymatic degradation in the gastrointestinal tract, and low permeability across the intestinal mucosa (3, 4). Therefore additional research is necessary to improve the oral bioavailability of these peptidergic analogs.
The results from a series of experiments investigating intestinal permeability implied that increases in permeability arose from the decrease in desolvation energy required to remove the hydrophilic molecule from the aqueous environment of the lumen into the lipophilic environment of the intestinal epithelial cell membrane (2). This finding is consistent with previous investigations which showed that the hydrogen binding ability of peptides affects their intestinal permeability (5). However, the above postulations are based on the modification of one GPIIb/IIIa peptidomimetic antagonist (2). More systematic investigations are required to study the factors that affect the intestinal permeability of RGD peptide analogs. For example, it has been shown that polar surface area has an inverse correlation with the transepithelial transport (6). The influence of other molecular parameters such as radius (7), geometry (8, 9), cross-sectional diameter (10), and volume (11) on transepithelial transport has also been reported.
Thus, the objective of this work was to model the influence of systematic methylation of RGD peptides on the conformationally dependent molecular parameters relevant to transepithelial transport.
MATERIALS AND METHODS
Materials
The Caco-2 cell line was purchased from American Type Culture Collection (ATCC), Rockville, MD. Dulbecco's modified eagle's medium (DMEM) and fetal bovine serum were purchased from JRH Biosciences, Lenexa, KS. Modified eagle's medium non-essential amino acids (100 x) solution, L-glutamine (200Mm), penicillin-streptomycin solution and trypsin-EDTA (0.05% trypsin, 0.53 mM EDTA) were from Gibco Laboratories, Life Technologies Inc., Grand Island, NY. Rat tail collagen was obtained from Collaborative Research Inc., Bedford, MA. Cluster dishes with 12 mm snapwells TM were obtained from Costar (Cambridge, MA). Parent RGD and the triethylammonium phosphate (TEAP) buffer were obtained from Novabiochem Inc., Houston, TX. The C-18 Nucleosil columns for HPLC analysis were obtained from Alltech Inc, Grand Island, NY. For the synthesis of RGD-r2 the following chemicals were used: Amino acids were obtained from Genzyme Corp, Cambridge MA, Wang resin from Calbiochem - Novabiochem Corp, San Diego CA, dimethylacetamide from EM Science, Gibbstown MA and dichloromethane from Burdick & Jackson, Muskegon MI. Triphenylphosphine, piperidine and triisopropylsilane were obtained from Aldrich Chemical, Milwaukee WI. Radiolabeled [1-14C] glycine-sarcosine (Gly-Sar; 55 Ci/mol) was obtained from American Radiolabeled Chemicals, Inc. (St. Louis, MO). 2-(N-morpholino) ethanesulfonic acid (MES) was purchased from Sigma Chemical Co. (St. Louis, MO).
Molecular modeling
Molecular modeling programs SYBYL (V 6.4), MACROMODEL, SPARTAN (V 5.0) and MAREA were used to investigate possible conformations of RGD and its methyl analogs based on their ionization state at pH 7.4. The analogs were all of the mono-, di-, and tri-methyl derivatives of RGD (Figure 1).
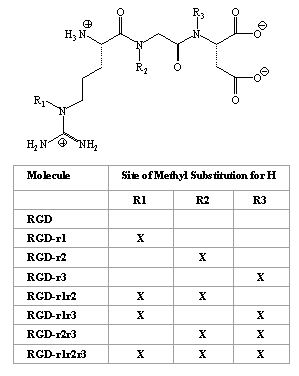
Figure 1: The basic chemical structure of arginine-glycine-aspartic acid (RGD) and its seven methyl substituted analogs under study
Silicon Graphics Indigo 2 computers at the National Center for Natural Products Research molecular modeling laboratory at the University of Mississippi were used for the modeling studies.
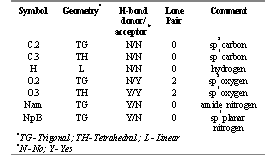
The initial structure of RGD and its methyl analogs as would be present at pH 7.4 were drawn using the biopolymer option in SYBYL. Atomic structural parameters such as atom and bond types (Table 1), the configuration of chiral centers, and the charge allocation were determined. Electrostatic point charges for the atoms were calculated within the program using two emperical methods. The s charges were calculated according to the method described by Gasteiger and Marsili (12). The p charges were calculated using the Hückel method (12). The two chiral centers were assigned the appropriate configuration. The Tripos Force Field parameters were used for the energy minimization calculations.
Table 1: Tripos force field atom types employed in the molecular modeling study of arginine-glycine-aspartic acid (RGD) analogs.
After an initial minimization, annealing was performed by heating the molecules to 700 K and then cooling to 300 K over a period of 1000 femto seconds (fs) for ten cycles. Following annealing, minimization was performed using the Powell descent series method. The minimization run was terminated when the gradient was less than 0.05 Kcal/mol. There were significant energy barriers to carbon-carbon and carbon-nitrogen bond rotation within the ring-like final conformation. Rotation about individual bonds carried a high potential energy penalty and therefore the minimized structures with the lowest potential energy were chosen for the study. Finally, each molecule was solvated with a water droplet using the appropriate solvation routine within SYBYL and minimized within that droplet. This final molecular structure was extracted from the water molecule and used for further calculations. It is noted that the solvated structures did not differ significantly in terms of geometry and potential energy with respect to the corresponding structures determined in vacuo .
Determination of structural parameters
SYBYL was used to determine the RGD H-bonding potential. This was calculated for each peptide according to Stein (13), where each amide N-H and carbonyl were assigned a value of 1, and each terminal amino and carboxyl a value of 2. Thus the total number of potential hydrogen bonds were obtained. The number of sites involved in intramolecular H-bond formation determined using SYBYL, were subtracted from the total number of potential sites, providing the total number of sites that would be available for H-bond formation with the hydrated polar heads of the lipid layer of the intestinal membrane. The distances between atoms involved in the hydrogen bond formation were measured with SYBYL to ascertain that the hydrogen bonds considered were strong enough to exist. The SYBYL Molcad module was used to study the lipophilic and electrostatic potential surfaces. Structures obtained in SYBYL were imported into Spartan version 5.0, which was used to calculate molecular surface areas, volumes, minimum and maximum cross-sectional diameters, and dipole moments. The conformations obtained using SYBYL were exported into Macromodel and were further analyzed for polar surface area using MAREA (6). The ACD predictive software of Advanced Chemistry Development (Toronto, Canada) was used to calculate the octanol-water partition coefficient for each of the molecules.
Caco-2 cell culture
Caco-2 cells were maintained at 37°C in Dulbecco's modified eagle's medium (DMEM), supplemented with fetal bovine serum (FBS), 1% non-essential amino acids, 1% L-glutamine and 100 U/ml penicillin and 100 m/ml streptomycin. Cells grown in T-flasks were split every week at a ratio of 1:3 before they reached confluency. The Caco-2 cells were plated at a density of 71 x 103 cells/transwell, previously coated with collagen. The cells were counted using a hematocytometer. The medium was changed on alternate days. After a period of 21 days the monolayers were used for the transport studies. The monolayers were not used after 30 days to ensure that the efflux mechanisms remained functional (14).
Determination of transepithelial electrical resistance (TEER)
The change in transepithelial resistance across the monolayers was studied. The resistance across the monolayers was measured every 24 h and the change in the values were recorded. The determinations were carried out at 37°C under open circuit conditions (15). The TEER value was calculated according to the following formula:
TEER ( W cm2 ) = (Total resistance - Blank resistance) ( W ) x Area (cm2 )
Typically, values between 150 and 250 ( W cm2) were observed.
In vitro transport studies
The Caco-2 cell monolayers were mounted on the diffusion chambers. The cells were bathed on both sides with 5 ml of bicarbonate-Ringer's solution (pH 7.4) supplemented with D-glucose to yield a final concentration of 40 mM and continously aerated with O 2 /CO 2 (95%/5%) at 37°C. After an initial equilibration the test agent was added. The samples were collected in triplicate at regular intervals and the volume taken was replenished with Kreb's-Ringer's buffer. Transport studies were conducted from apical to basolateral, basolateral to apical, and across a pH gradient and at lower pH values. In order to carry out transport experiments across a pH gradient or at a lower pH, 10 mM 2-[N-morpholino]ethanesulphonic acid (MES) was added to the bicarbonate-Ringer's buffer and the pH was adjusted to 6.0 using hydrochloric acid. The transport of the dipeptide glycine-sarcosine (Gly-Sar) was studied in the presence of RGD in order to examine a possible inteference of the tripeptide with gly-sar transport.
Synthesis of the RGD analog
RGD peptide synthesis consisted of the following procedures. The initial step involved the attachment of the first amino acid to a polymeric solid support. The second step was the chain elongation by a solid phase peptide synthesis. In the third step the peptide was cleaved off the polymer support followed by purification. Peptides were synthesised from the C-terminus to the N-terminus. The C-terminal amino acid was attached to polyethylene glycol polystyrene resin beads using the Mitsunobu reaction. The amino acid, triphenylphosphine, diethylazodicarboxylate and the resin beads were combined and stirred in a one-pot reaction overnight at room temperature. The resin was filtered, washed and dried. Solid phase synthesis was carried out in a glass reaction vessel by treating the resin with 20% piperidine in dimethylacetamide to remove the N-terminal protecting group. The resin was later washed several times with dimethylacetamide. The next amino acid was dissolved with 2-(1H-benzotriazole-1-y1)-1,1,3,3-tetramethylamonium hexafluorophosphate, di-isopropylethylamine in dimethylacetamide and dichloromethane. This was added to the resin and mixed for 1-2 hours at room temperature. The resin was later washed several times with dimethylacetamide and the coupling was tested for the completeness of reaction. These series of steps were repeated and carried out for each amino acid in the chain until it was complete. The final protecting group was removed and the peptide-resin was washed, filtered and dried. The peptide was cleaved off the resin by treating it with 95% trifluoroacetic acid/5% triisopropylsilane for one hour. Next the solution was evaporated to dryness under vacuum. Diethyl ether was added and the ether was filtered away leaving the precipitated peptide and resin. The peptide was extracted off the resin with 10% aqueous acetic acid, acetonitrile and distilled water. The solution was then lyophilized. The crude material was purified using reverse phase chromatography. Fractions were collected and lyophilized. The peptide purity was assayed by HPLC and LC/MS.
Sample analysis
The analysis of the samples obtained from the transport studies of RGD analogs was carried out using a Hewlett Packard 1090 HPLC instrument, San Diego, CA and an isocratic, 0.05 M triethylammonium phosphate (TEAP) buffer at pH 2.25 as the mobile phase. The column used was a Nucleosil C-18 column (25 x 4.6 cm) from Alltech, Inc, Grand Island, NY (100 l aliquots of the transport samples obtained from the donor and receiver sides were injected). The flow rate was maintained at 0.5 ml/min. The compounds were analyzed using a UV detector at a wavelength of 210 nm. Permeabilities were estimated from the slope of the cumulative amount versus time normalized for the initial concentration and the area of the filter. The radioactive gly-sar samples were placed in scintillation vials, mixed with scintillation cocktail (Ready Safe Beckman Instruments, Fullerton, CA) and counted in a scintillation counter using an external standardization method.
pKa and lipophilicity studies
The calculated values for pKa, Log P (octanol/water partition of the neutral species), and Log D (octanol/buffer partition at indicated pH of ionized and unionized molecules) were determined using ACD/Log P and ACD/Log D software from Advanced Chemistry Development, Toronto, Canada. The experimental determination of these parameters were performed using a potentiometric method for RGD and RGD-r2 (16). An instrument from Pion/Sirius was used for the pKa and cLog P (calculated Log P) determinations. Measurements were performed in duplicate. One mg of the RGD analog was used under a constant flow of argon. For maintaining the pH and titrating between the pH range of 1.8 to 12, potassium hydroxide and hydrochloric acid were used. The pKa values were obtained using the pKa and Log P analyser software program. For the Log P and Log D determinations the shift in these pKa values in the presence of octanol saturated with water were also measured. The program also gave the percentage distribution of each ionic species at various pH ranges.
RESULTS AND DISCUSSION
Hydrogen bonding potential
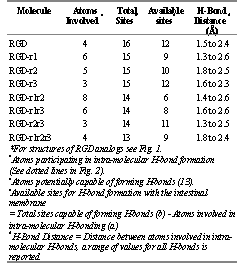
Results obtained from the molecular modeling studies suggest that RGD and its methyl analogs (Figure 1) have a significant H-bonding potential, both intra- and intermolecular (Figure 2, Table 2). Intermolecular H-bond formation may occur between the molecules and the hydrated polar heads of the lipid bilayer of the intestinal membrane. Initially we hypothesized that with the addition of a methyl group, one potential hydrogen bonding site would be eliminated. Based on this assumption, the total number of sites would have been 16 for the non-substituted RGD, 15 for the mono-substituted analogs, 14 for the di-substituted analogs and 13 for the tri-substituted analog (Figure 1).
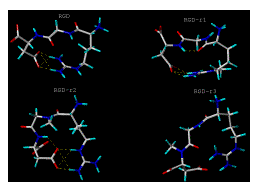
Figure 2: [Panel(a)]. Computer generated lowest energy conformations of RGD, RGD-r1, RGD-r2 and RGD-r3 analogs using SYBYL after annealing, minimization, solvation and extraction. The pictures show the number and sites involved in potential intramolecular H-bond formation. Dotted lines indicate intra-molecular H-bonds. For key to structures see Figure 1.
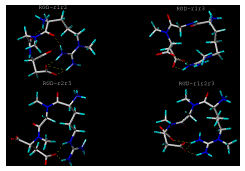
Figure 2: [Panel(b)]. Computer generated lowest energy conformations of RGD-r1r2, RGD-r1r3, RGD-r2r3 and RGD-r1r2r3 analogs using SYBYL after annealing, minimization, solvation and extraction. The pictures show the number and sites involved in potential intramolecular H-bond formation. Dotted lines indicate intra-molecular H-bonds. For key to structures see Figure 1.
Table 2: Hydrogen bonding ability of arginine-glycine-aspartic acid (RGD) analogs*
However the molecular modeling results indicate that not all these sites will be available for H-bonding with the intestinal membrane, since many of the sites in the RGD analogs may potentially be involed in intramolecular H-bond formation (Figure 2). The total number of sites or atoms involved in the intramolecular H-bond formations were found to depend not only on the degree of substitution, but also on the site of methyl substitution and the conformation of the molecule. Results indicate that the mono-substituted RGD-r1 has 9 sites potentially available for H-bonding with surrounding intestinal fluid in comparison to 10 and 12 sites available for the other mono-substituted analogs RGD-r2 and RGD-r3, respectively (Table 2). Similarly the di-substituted analog, RGD-r1r2 has 6 available sites for hydrogen bonding with the intestinal membrane in comparison to 8 and 11 sites available for RGD-r1r3 and RGD-r2r3, respectively. The analog with three methyl subsitutions, i.e., RGD-r1r2r3, has 9 sites available for H-bonding with the intestinal membrane.
The strength of H-bonds was investigated by computing the distance between atoms involved in H-bond formation. The smaller the distance between the atoms, the greater would be the strength of the H-bonds. The interatomic distances involved in H-bonding were found to range between 1.5 and 2.6 Å (Table 2). It was concluded that the H-bonds had sufficient interaction potential and were relevant to the study.
Structural comparison of RGD peptidomimetic analogs have suggested that the methylation of amide bonds may have resulted in the increased intestinal permeability and therefore increased bioavailability of the methylated compounds (2). This suggestion was based on previous investigations by Burton et al. (17) who showed that the general hydrogen bonding ability of peptides affects their intestinal permeability. However, the postulation that the methylation of amide bonds may have resulted in the increased intestinal permeability of the RGD peptidomimetic agents was based only on the modification of one GPIIb/IIIa peptidomimetic antagonist (2). Therefore more systematic investigations are required to study the factors that affect the intestinal permeability of RGD peptide analogs. The RGD analogs under study were modeled to demonstrate a systematic increase in the number and site of methyl substitution (Figure 1). Our data suggest that the hydrogen bonding ability of RGD analogs did not directly correlate with a successive increase in the number of methyl substitutions. The H-bonding ability was shown to be a function of the solution conformation of the molecules which was influenced by both the number and the site of methyl substitution.
Molecular geometry
The variation in molecular surface area was both a function of the site as well as the number of methyl substitution, ranging between 340 Å2 and 416 Å2 (Figure 3). The molecular volume varied between 358 Å3 and 416 Å3. An increase in the number of methyl substitution was accompanied by an increase in molecular volume (Figure 3). No correlation was observed between the molecular weight of the peptide and the molecular surface area, however in general an increase in molecular weight with successive methylations resulted in an increase in molecular volume.
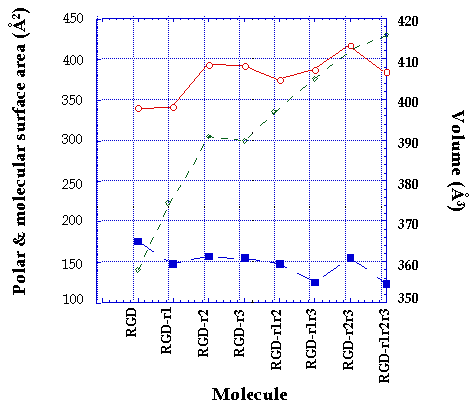
Figure 3: The effect of the number and site of methyl substitutions of RGD analogs on the calculated molecular volume (à), total molecular surface area (o) and polar surface area (n). For key to structures see Figure 1.
The polar surface area (PSA) for each of the analogs was obtained using MAREA software (Figure 3). The PSA varied between 123 Å2 and 175 Å2. The lowest polar surface area was found to be for the tri-substituted RGD-r1r2r3 and the highest was for the parent non-substituted RGD. Slight changes in polar surface area were observed with a change in the site of methyl substitution.
Consistent with reports in the literature (6, 18), a general correlation was obtained between polar surface area and the hydrogen bonding ability of the RGD analogs (Figure 4). It has been shown that polar surface area, i.e., the area occupied by nitrogen, oxygen and hydrogen atoms attached to the heteroatoms, is representative of the compounds hydrogen bonding ability (6, 19). It should be noted that the total number of available sites present in each analog is more important for the study, though as expected it does give a correlation with polar surface area.
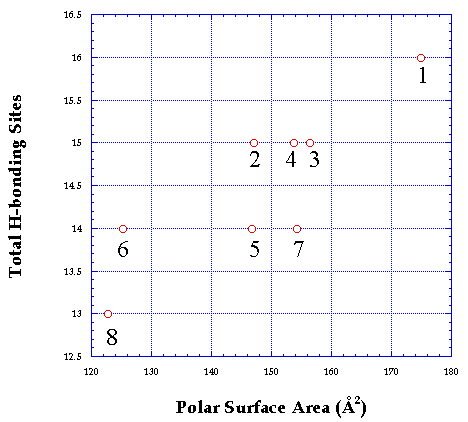
Figure 4: Correlation between polar surface area and total hydrogen bonding potential of the RGD analogs. (1) RGD, (2) RGD-r1, (3) RGD-r2, (4) RGD-r3, (5) RGD-r1r2, (6) RGD-r1r3, (7) RGD-r2r3, (8) RGD-r1r2r3. For key to structures see Figure 1.
Successive methyl substitution of the RGD analogs reduced the polar surface area, however the extent of decrease varied with the site of methyl substitution. It has been shown that compounds with polar surface area greater than 120 Å2 are poorly absorbed by the passive transcellular route (18, 19). Polar surface areas of the RGD analogs under study are generally higher than 120 Å2, therefore no significant transport of the analogs was predicted.
The maximum and minimum cross-sectional diameters were determined using SYBYL (Table 3). Maximum cross-sectional diameter varied between 9.2 Å and 11.8 Å, which was greater than the value of 8 Å obtained by the equivalent pore theory for the paracellular space (20), therefore supporting the prediction of low transepithelial transport of the analogs. The minimum cross-sectional diameter varied between 4.9 Å to 7.9 Å. It was observed that an increase in the number of methyl substitutions did not directly correlate with an increase in the diameters of the analogs (Table 3).
Table 3: Cross-sectional diameters, calculated log P values (ACD) and dipole moments of arginine-glycine-aspartic acid (RGD) analogs
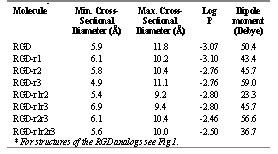
The SYBYL Molcad module was used to study the lipophilic and electrostatic potential surfaces of the RGD and its methyl analogs (Figure 5). The molecular surface study indicated that the most lipophilic and neutral part of the molecule, which in most likelihood would approach the intestinal membrane, will constitute the maximum cross-sectional diameter which will thus be an important parameter to consider in transport (21).
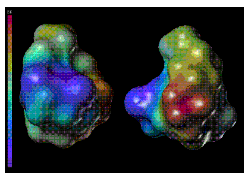
Figure 5: [Panel(a)]. Computer generated structures of RGD (left), and RGD-r1r2 (right) analogs representing the calculated electrostatic (red is most electropositive and blue is most electronegative) potential surfaces of the molecules. For key to structures see Figure 1.
The dipole moment, which shows the extent of polarity of a compound, was determined using Spartan and was found to be the lowest for RGD-r1r2 and highest for RGD-r3 (Table 3). The values obtained further support the importance of conformation in the determination of various structural parameters such as dipole moment and cross-sectional diameter.
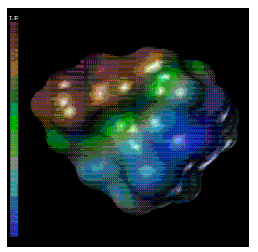
Figure 5: [Panel(b)] Computer generated structure of RGD-r1r2 analog representing the calculated lipophilic (red surface is most lipophilic and blue is the least lipophilic) potential surface of the molecule. For key to structures see Figure 1.
The importance of molecular geometry and polarity on the transepithelial transport of molecules has been emphasized by various investigators (8, 9). Permeability has been correlated with geometrical parameters such as molecular volume (11) maximum and minimum cross-sectional diameters (7, 10) and polar surface areas (18, 19). Since the successive methylation of the RGD analogs under study was shown to influence the conformation of the compounds, we studied the effect of such change on their geometrical parameters. No significant change was observed for the total molecular surface area indicating that methyl substitution does not significantly affect these geometrical parameters. However an increase in methyl substitution correlated with a corresponding increase in molecular volume (Figure 3). Polar surface area did correlate with H-bonding ability of the compounds. Our results also indicate that in addition to molecular geometry, the orientation of the molecule towards the intestinal membrane may be influenced by the electrostatic and the lipophilic potential of the compounds (Figure 5).
Log P & pKa studies
We calculated cLog P using the ACD software and found very little variations based on methyl substitution (Table 3). Typically, cLog P values were less than -2.46 for all examined RGD analogs. We also determined Log P values of RGD and RGD-r2 experimentally and found values less than -2, confirming cLog P estimations. In accordance with Log D values obtained using ACD, the potentiometric method used over the pH range of 1.8 to 12 could not provide any Log D value higher than -2. The pKa values obtained for RGD were 2.8, 4.0, and 6.8. The values obtained for RGD-r2 were 3.3, 4.2, and 7.1. The pKa values of these compounds aided us in understanding the distribution of each ionic species. The dipole moment of RGD and its methyl analogs was determined in order to understand the polarity of the analogs and also to understand the distribution of charge in the molecule, which would help in comparing the polarity of the analogs (Table 3). It has been shown that the polarity of the compound is a key determinant of lipophilicity (22). In the present study the dipole moment of the analogs was found to be the lowest for RGD-r1r2 (23.3 Debye) and the highest for RGD-r3 (59.0 Debye). The difference in values obtained for dipole moments indicate the importance of the arrangement of atoms in conformational space. The Log P values were calculated, as lipophilicity influences transepthelial transport especially through the transcellular route (23, 24). Lipophilicity is commonly expressed as the apparent partition coefficient between the organic and the aqueous phases. Unlike Log D values which are pH dependent, Log P values are pH independent. Log D is a measure of all species including ionic whereas Log P is a measure of neutral species only (25). It is noted that with one methyl substitution the pKa values were not significantly influenced.
In vitro transport study
Based on the data discussed above, i.e., the extensive hydrogen bonding (6-12 sites), large polar surface area (> 120 Å2 ), low Log P values (< -2) and high maximum cross-sectional diameters (> 8 Å) of the analogs, it was predicted that limited transport of the RGD analogs would take place. In order to validate these predictions, transport experiments for RGD and RGD-r2 as model compounds were conducted using Caco-2 cell monolayers. No detectable transport was observed for RGD or the mono-substituted RGD-r2 analog. The detection limit for RGD and RGD-r2 was approximately 1 mg/ml. The stability of the peptide analogs during the course of the transport studies was verified by analysing the samples in the apical chambers at the conclusion of each experiment. We did not observe an appreciable loss of peptide concentration, suggesting that peptide stability was not a factor for lack of transport.
In order to examine whether the transport of these RGD peptides could be mediated by PepT1, transport was assessed in the absence (pH 7.4) or presence (pH 6.0) of an apical proton gradient across Caco-2 monolayers, which are known to express PepT (26). As a positive control we included Gly-Sar in our studies. As expected Gly-Sar transport increased from 4.2 x 10-6 to 6.2 x 10-6 cm/sec in the presence of a pH gradient. Our pH 6.0/7.4 Gly-Sar flux ratio of 1.48 is in good agreement with reports by Thwaites and co-workers (27) who calculated a ratio of 1.57 for the same substrate. A significant finding, however, was the reversal of the augmented transport of Gly-Sar in the presence of RGD from 6.2 x 10-6 to 4.0 x 10-6 cm/sec indicating that RGD was either competitively inhibiting the binding of Gly-Sar to PepT1 and/or interfering with the proton transport across the apical membrane (Figure 6).
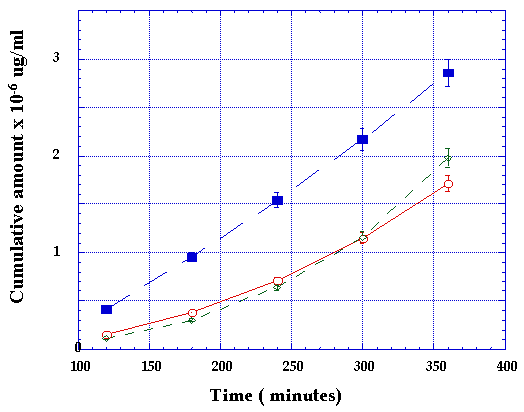
Figure 6: Transepithelial transport of gly-sar at pH 6 (n), gly-sar at pH 7.4 (à) and Gly-sar in the presence of RGD at pH 6 (o).
SUMMARY AND CONCLUSION
Results of our study point out several issues that are important in the design considerations of new peptidomemitic RGD analogs with enhanced oral bioavailability. It was shown that the hydrogen bonding ability of the compounds appears to be a function of both the site and the number of methyl substition. An increase in hydrogen bonding ability was correlated with a general increase in polar surface area. Molecular geometry was shown to be influenced by both the site and the number of methyl substitution. Molecular volume showed a direct correlation with the number of methyl substitutions. We acknowledge that the linear RGD analogs studied in the present work may not be the active forms of the compounds and many cyclic analogs have shown antithrombotic activity. However since a common basic component of the peptidergic GPIIb/IIIa antagonists is the RGD sequence, we chose the linear template and its analogs as a building block for a systematic correlation of structure-transport relationships. Future studies may include the correlation of the structural parameters of bioactive RGD peptidomimetic analogs with their transepithelial transport. It is anticipated that such systematic studies will aid in the design of new compounds or delivery systems that aim at increasing the oral bioavailability of GPIIb/IIIa antagonists for chronic antiplatelet therapy.
ACKNOWLEDGMENTS
We would like to thank Genentech Inc. (South San Francisco, CA) for providing Rudraksh Sharan the opportunity to work on this project through the summer internship program, Drs. Steven Bishop, Martin Struble, and Dean Artis at Genentech, Inc., for the synthesis of the RGD analogs, and Dr. Per Artursson and co-workers at the University of Uppsala, Division of Pharmaceutics (Uppsala, Sweden) for having provided us with the MAREA software (Palm et al. 1997).
REFERENCES
Nichols, A. J., Ruffolo, R. R., Huffman, W. F., Poste, G., and Samanen, J., Development of GPIIb/IIIa antagonists as antithrombotic drugs. Trends in Pharmacol. Sci., 13:413-417, 1992.
Samanen, J., Wilson, G., Smith, P. L., Lee, C. P., Bondinell, W., Ku, T., Rhodes, G., and Nichols, A, Chemical approaches to improve the oral bioavailability of peptidergic molecules. J. Pharm. Pharmacol., 48:119-135, 1996.
Smith, P. L., Wall, D. A., Gochoco, C. H., and Wilson, G., Oral absorption of peptides and proteins. Adv. Drug Del. Rev., 8:253-289, 1992.
Lee, V. H. L., Dodda-Kashi, S., Grass, G. M. and Rubas, W., Oral route of peptide and protein drug delivery, in Lee VHL (ed) Peptide and Protein Drug Delivery. 2nd ed., Marcel Dekker, INC, New York, pp. 691-740, 1991.
Burton, P. S., Conradi, R. A. and Hilgers, A. R., Transcellular mechanism of peptide and protein absorption: passive aspects. Adv. Drug Del. Rev., 7:365-386, 1991.
Palm, K., Stenberg, P., Luthman, K., and Artursson, P., Polar molecular surface properties predict the intestinal absorption of drugs in humans. Pharm. Res., 14:568-571, 1997.
Sawada, T., Tomita, M., Hayashi, M., and Awazu, S., Paracellular channel characterized by non-electrolyte permeation through the colonic membrane of the rat. J. Pharmacobio. Dyn., 12:634-639, 1989.
Ghandehari, H., Smith, P. L., Ellens, H., Yeh, P-Y. and Kopecek, J., Size dependent permeability of hydrophilic probes across rabbit distal colonic mucosa. J. Pharmacol. Exp. Ther., 280, 747-753, 1997.
Hollander, D., Ricketts, D. and Boyd, C. A. R., Importance of probe molecular geometry in determining intestinal permeability. Nutr. Ther., 2:35-38, 1988.
Lane, M. E., Caitriona, M. and Corrigan, O. I., The relationship between rat intestinal permeability and hydrophilic probe size. Pharm. Res., 13:1554-1558, 1996.
Hamilton, I., Rothwell, J., Archer, D. and Axon, A. T. R. Permeability of the rat small intestine to carbohydrate probe molecules. Clin. Sci., 73:189-196, 1987.
Gasteiger, J. and Marsili, M., Iterative partial equalization of orbital electronegativity-A rapid access to atomic charges. Tetrahedron, 36: 3219-3228, 1980.
Stein, W. D., The movement of molecules across cell membranes. Academic Press, New York, USA, pp.65-91, 1967.
Delie, F. and Rubas, W., A human colonic cell line sharing similarities with enterocytes as a model to examine oral absorption: advantages and limitations of the Caco-2 model. Cri. Rev. in Ther Drug Carr. Sys., 14:221-286, 1997.
Rubas, W., Cromwell, M. E. M., Shahrokh, Z., Villagran, J., Nguyen, T. -N.Wellton, N., Nguyen, T. H., and Mrsny, R. J., Flux measurements across Caco-2 monolayers may predict transport in human large intestinal tissue. J. Pharm. Sci., 85:165-169, 1996.
Leo, A., Hansch, C. and Elkins, D., Partition coefficients and their uses. Chem. Rev., 71:525-554, 1971.
Burton, P. S., Conradi, R. A., Hilgers, A. R., Ho, N. F. H. and Maggiora, L. L., The relationship between peptide structure and transport across epithelial cell monolayers. J. Contr. Rel., 19:87-98, 1992.
Palm, K., Luthman, K., Ungell, A., Strandlund, G., and Artursson, P. Correlation of drug absorption with molecular surface properties. J. Pharm. Sci., 85:32-39, 1996.
Stenberg, P., Luthman, K., and Artursson, P., Prediction of membrane permeability to peptides from calculated dynamic molecular surface properties. Pharm. Res., 16:205-212, 1999.
Solomon, A. K. Characterization of biological membranes by equivalent pores. J. Gen. Physiol., 51:335S-364S, 1968.
Pauletti, G. M., Okumu, F. W., and Borchardt, R. T., Effect of size and charge on the passive diffusion of peptides across Caco-2 cell monolayers via the paracellular pathway. Pharm. Res., 14:164-168, 1997.
Norinder, U., Österberg, T., and Artursson, P., Theoretical calculation and prediction of intestinal absorption of drugs in humans using MolSurf parametrization and PLS statistics. J. Pharm. Sci., 8:49-56., 1999
Wils, P., Warnery, A., Phung-Ba, V., Legrain, S., and Scherman, D., High lipophilicity decreases drug transport across intestinal epithelial cells. J. Pharmacol. Exp. Ther., 269:654-658, 1994.
Artursson, P. and Magnusson, C. J., Epithelial transport of drugs in cell culture: II. Effect of extracellular calcium concentration on the paracellular transport of drugs of different lipophilicities across monolayers of human intestinal epithelial (Caco-2) cells. J. Pharm. Sci., 79:595-600, 1990.
Rubas, W., Cromwell, M. E. M., The effect of chemical modifications on octanol/water partition (Log D) and permeabilities across Caco-2 monolayers, Adv. Drug. Del. Rev., 23:157-162, 1997.
Oh, D. M., Uptake of a dipeptide by the dipeptide transporter in the HT-29 intestinal cells. J. Kor. Pharm. Sci., 25:137-143, 1995.
Thwaites, D. T., Brown, C. D. A., Hirst, B. H., Simmons, N. L., Transepithelial glycylsarcosine transport in intestinal Caco-2 cells mediated by expression of H+-coupled carriers at both apical and basal membranes. J. Biol. Chem., 268:7640-7642, 1993.
Corresponding Author: Hamidreza Ghandehari, Department of Pharmaceutical Sciences, University of Maryland School of Pharmacy, Baltimore, MD 21201, USA,
hghandeh@rx.umaryland.edu
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps