J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 4(1):1-6, 2001
Pharmacokinetics of Celecoxib in the Presence and Absence of Interferon-Induced Acute Inflammation in the Rat: Application of a Novel HPLC Assay
Micheal S. Guirguis, Saeed Sattari, Fakhreddin Jamali1
Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, CanadaReceived December 1st, 2000, Revised February 15th, 2001, Accepted February 16th, 2001
PDF version for printing
ABSTRACT
Purpose. Celecoxib (CEL) is a relatively new cyclooxygenase-2 specific inhibitor nonsteroidal anti-inflammatory drug with low incidents of the toxic side effects. We developed and validated an HPLC assay for CEL and delineated pharmacokinetics of the drug in the rat in the presence and absence of inflammation.
Methods. Rat plasma (0.1 mL plasma) was spiked with CEL and ibuprofen as internal standard. The solution was acidified and constituents were extracted with isooctane-isopropanol (95:5). The organic solvent was separated, evaporated and the residue was dissolved in the HPLC mobile phase [acetonitrile-water-acetic acid-triethylamine (47:53:0.1:0.03)]. The HPLC system consisted of an auto-injector, an isocratic pump, a 10 cm C18 analytical column packed with 5-mm of reversed-phase particles, a UV detector set at 254 nm, and an integrator. Control adult male Sprague-Dawley rats were dosed with CEL [5 mg/kg i.v. (n=8), p.o. (n=6) or i.p. (n=3)]. Acute inflammation was brought about by two (12 and 1 h pre-CEL) s.c. injection of 50,000IU/200mL interferona2a. Inflamed rats (n=6) received 5 mg oral CEL. Serial blood samples were collected via a inserted catheter at the right jugular vein, and plasma samples were analyzed for CEL.
Results. The assay yielded linear response within the examined ranges of 20-1000 ng/mL and 1-100 mg/mL (r2>0.99) with an extraction efficiency of >70%, intra- and inter-day variability of <10% and accuracy of >90%. In control rats, CEL had an oral bioavailability of 0.59 due mainly to presystemic hepatic metabolism. A multi-compartmental disposition kinetics with an average terminal t1/2 of 2.8 ± 0.7 h, and volume of distribution of 2.3 ± 0.6 L/kg were found. Acute inflammation had no significant effect on the pharmacokinetics of CEL, although a trend towards increased plasma concentration was noticed.
Conclusions. The validated assay has sufficient accuracy and precision for pharmacokinetic studies of CEL in the rat. The lack of change in CEL pharmacokinetics after acute inflammation maybe due to 1) insensitivity of its metabolic system to the acknowledged inhibitory effect of inflammation, and/or 2) the relatively low pre-systemic metabolism of the drug.
Introduction
Celecoxib (CEL), a member of the new generation of non-steroidal anti-inflammatory drugs (NSAIDs) (Figure 1.), specifically acts on cyclooxygenase-2 (COX-2), the main cyclooxygenase isomer expressed during inflammation (1-4).
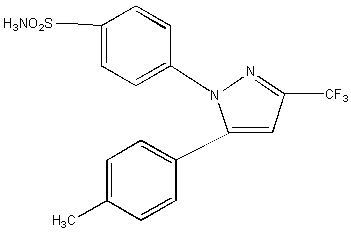
Figure 1: The Chemical Structure of CEL.
Previous generations of NSAIDs inhibit both cyclooxygenase -1 (COX-1) and COX-2. COX-1, is constitutively expressed and is associated with maintenance of the cell (e.g., mucosal lining of gut integrity) hence, its inhibition results in clinically significant toxicity(5, 6). This toxicity may be prevented with the introduction of NSAIDs with high selectivity in COX-2 inhibition(4-6). At present, CEL is primarily used in the treatment of pain and inflammation associated with rheumatoid arthritis and osteoarthritis. Inflammatory conditions such as rheumatoid arthritis and administration of interferon have been reported to significantly inhibit hepatic metabolism of many drugs (7-9). Therefore, we developed an assay suitable for delineation of pharmacokinetics of CEL in the rat and studied the effect of acute inflammation caused by administration of interferona2a. The reported high performance liquid chromatography (HPLC) assays (10) use solid-liquid extraction procedures. Herein, we report an HPLC assay, which involves a convenient and cost-effective liquid-liquid extraction method.
Methods and Materials
Chemicals
Celecoxib was extracted from Celebrex 100 mg Capsules (Searle MI, USA). Racemate ibuprofen was obtained as a gift from Upjohn Canada (Don Mills, Canada). Isooctane and isopropanol (Assurance grade) were purchased from BDH Chemicals Canada (Edmonton, Canada). HPLC grade acetonitrile and methanol as well as analytical grade triethylamine, acetic acid and sulfuric acid were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Interfrona2a (Referon-A) was from Hoffmann-La Roche Ltd (Mississauga, Ontario, Canada).
Assay Preparation
The HPLC system (Shimadzu, Japan) consisted of a Sil-9A model autoinjector, a SPD-6A model variable UV spectrophotometer detector set at 254 nm and a CR601 model Chromatopac integrator, and a Waters 6000 A model HPLC pump (Waters, Mississauga, Canada). The mobile phase, which was filtered through a 0.45 mm nylon filter before use, consisted of acetonitrile-water-acetic acid-triethylamine (47:53:0.1:0.03) and was pumped at a flow rate of 1 ml/min. A 10 cm x 4.6 mm I.D. C18 analytical column packed with 5 mm reversed-phase particles (Phenomenex, Torrance, CA, USA) and a Novo-Pak C8 guard column (Waters, Millipore Corp., Milford, MA, USA) were used for separation.
Celecoxib powder were prepared from the contents of 20 capsules, which were suspended in 50 mL of HPLC grade methanol, filtered, allowed to evaporate, and dissolved in and recrystallized from acetonitrile. The identity and purity of the CEL was determined utilizing melting point, differential scanning calorimetry, and nuclear magnetic resonance (data not shown). Standard solutions were prepared by adding 100 mL of ibuprofen stock solution (100 mg/mL) and specific volumes of CEL stocks solutions (1 mg and 100 mg/mL) to 100 ml of rat blank plasma to make the final concentrations of 20 ng/mL to 100 mg/mL. Two calibration curves with different concentration ranges were prepared. The low range curve included solutions containing 20, 50, 100, 250, 500, 1000 ng/mL and the high range curve consisted covered solutions of 1.0, 2.5, 5.0, 25, and 100 mg/mL of CEL. To each spiked standard solution was added 0.2 ml of 0.6 M H2SO4 and 5 ml of isooctane-isopropanol (95:5). The tubes were vortex-mixed for 30 s and centrifuged at 2500 g for three min. The organic layer was transferred to clean tubes and evaporated (Savant Speed Vac concentrator-evaporator, Emerson Instruments, Scarborough, Canada). The residue was reconstituted in 200 mL of mobile phase and aliquots of 150 μL were injected to the chromatographic system.
The method's extraction efficiency was determined for concentrations of 50, 100 and 250 ng/mL. The peak areas of the extracted plasma samples were compared with the peak areas obtained after direct injection of 150 mL of solutions containing 50, 100 and 250 ng/mL CEL.
Accuracy was determined by examining calibration curves prepared in triplicate on three consecutive days.
Pharmacokinetic Experiments
Male Sprague-Dawley rats (250-300 g) were anesthetized using pentobarbital (65 mg/kg i.p. ) and a silastic catheter was inserted into the right jugular vein. Animals were allowed to recover for 24 h before dosing. On the day of the experiment animals were placed in metabolic cages and dosed with 5 mg/kg i.v. (n=8), p.o . (n=6) or i.p . (n=3) of CEL. Serial blood samples (200 mL) were collected. The catheter was flushed with 200 mL of saline after each sample collection. Samples were stored at -20° until analyzed. The effect of inflammation was assessed by dosing animals (n=6) with interferona2a (INF, 200 mL s.c .; is saline-diluted to a concentration of 5x 104iu/200 mL immediately before use) 12 and 1 h before administration of CEL . Control rats were dosed with 200 mL s.c. normal saline.
Data Analysis
Accuracy was expressed as the mean % error, [(mean measured concentration - expected concentration)]x100. Precision was expressed as percent coefficient of variation [(standard deviation/mean)x100].
Areas under the plasma concentration versus time curves (AUC) were calculated using the linear trapezoidal method. Elimination rate constants (b) were calculated for each individual rat from the slope of the regression line of the log-linear terminal elimination phase of the concentration-time curves using at least 3 data points. The oral clearance (CLoral) was calculated from Dose/AUC(0-¥). Volume of distribution was calculated from CL/b. Bioavailability (F) was calculated from AUCoral or AUC i.p /AUC i.v. . The significance of the difference between any two groups was tested using paired two-tailed Students t-test. Determination of significance of differences between multiple groups was done using ANOVA followed by the Duncan New Multiple Range test. The level of significance was set at 0.05. Data are expressed as mean ± standard deviation.
Results
Assay Validation
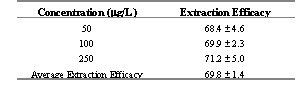
Ibuprofen and CEL were eluted 8 and 11 min following injection into HPLC, respectively. The peaks were resolved with no interfering peaks (Figure 2). Extraction efficiency was found to be 69.8 % (Table 1).

Figure 2: Representative chromatographs of: A) blank rat plasma, B) plasma spiked with 100 ng/mL ibuprofen (peak 1) and 100 ng/mL CEL (peak 2,) and, C) plasma sample from a rat 4 h after administration of 5 mg/kg, the concentration of 2 is equivalent to 151 ng/mL.
Table 1: Extraction efficacy of CEL from rat plasma at various concentrations, mean ± standard deviation (n=3)
The relationship between the area ratio of CEL to internal standard versus concentration was linear (r2<0.99) for both low and high range calibration curves. Intra- and inter-day variability did not exceed 10% with less that 10% error throughout the examined range (Table 2 and 3). The limit of quantification of the assay was, therefore, set at 20 ng/mL of CEL based on 0.1 mL rat plasma sample.
Table 2: Within-day precision and accuracy for the CEL assay in rat plasma.
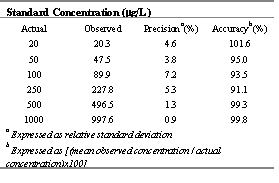
Table 3: Intra-day variability of the CEL assay over three consecutive days in rat plasma.
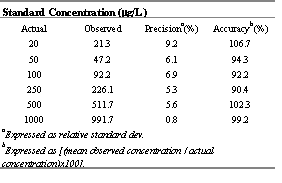
Table 4. Mean (± standard deviation) pharmacokinetic indices of CEL in control rats after various routes of administration and after p.o. administration in interferon-treated rats of 5 mg/kg.

Celecoxib pharmacokinetics
Following i.v. administration of CEL, the concentration-time curve of CEL exhibited a multi-compartment pattern with a steep initial declines followed by a log-linear terminal phase with a t1/2 of 3 ± 1 h (Figure 4).
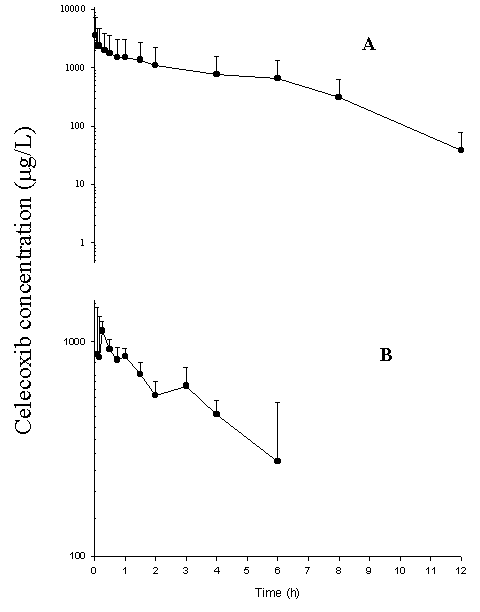
Figure 3: Concentration-time curve following (A) 5mg/kg CEL administered i.v. (n=8) and (B) i.p. (n=3). Data presented as mean ± standard deviation
CEL has a relatively large volume of distribution and a medium to low hepatic extraction (Table 4). Numerical differences were found in AUC values when CEL was administered through different routes with the greatest and smallest values after i.v. and p.o. , respectively (Table 4). These differences, however, were significant only between the i.v. and other routs.
Interferon administration was associated with significant increases in nitrite, a stable metabolite of nitric oxide, and segmented neutrophil indicating inflammation (11). Interferon therapy did not significantly alter pharmacokinetics of CEL although a trend toward increased AUC was observed (Table 4 and Figure 4).
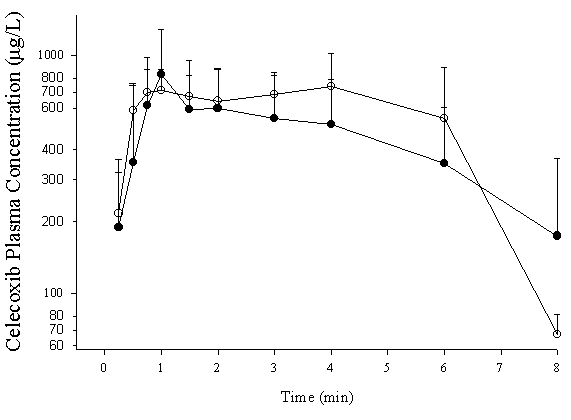
Figure 4: Concentration-time curve for CEL administrated orally (5mg/kg) to control (·) and interferon-treated (o) rats. Data presented as mean ± standard deviation (n=6/group).
Discussion
The present assay is sensitive, reproducible and convenient. The liquid-liquid extraction method used is more convenient and cost-effective than the solid-phase extraction procedure utilized by others.(10, 12, 13). This method provides a sensitivity of at least 20 ng/mL based on 100 mL sample volume, which is suitable for pharmacokinetic studies of CEL and is comparable with the assays reported by others (10). In a preliminary experiment we confirmed that the assay can also be used to determine CEL in human plasma.
Our basic pharmacokinetic indices consistent with values reported previously (12). In addition, a comparison of the AUC values following administration through different routes indicates that the drug is cleared mainly by the liver with the possibility of some metabolism and/or incomplete absorption taking place in the gut (AUC: i.v.>i.p.>p.o. ); i.e., the i.v. dose which by passes first pass metabolism has the greatest AUC while the p.o. dose which undergoes both gut and liver first-pass clearance yielded the smallest AUC. The involvement of the gut, however, is not certain since the numerical difference between the AUC of p.o . and i.p. routes did not indicate significance. Pharmacokinetic data following i.v. administration (Table 4) suggest a CLTB of 10 mL/min/kg for CEL.
Considering the significant contribution of the hepatic metabolism in clearance of CEL and the rat hepatic blood flow of 65 mL/min/kg, the drug may be classified as one with a low hepatic extraction. The relatively large volume of distribution of approximately 2 L/kg (Table 4) suggests extensive deep tissue binding and/or distribution. These findings in the rat suggest somewhat similar disposition kinetics to those reported in humans (14).
Both acute inflammation induced by interferon (11) and chronic arthritis (15) are reported to reduce clearance of drugs. This effect is mainly observed with drugs that are cleared in the liver. The extent of this inhibition increases with the efficiency of the hepatic metabolism; i.e., drugs with high hepatic extraction such as propranolol (16, 17) and verapamil (11, 15) are more affected than those with intermediate or low extraction ratio such as hyroxychloroquine (18). The diminishing effect of inflammation on clearance of drugs is attributed to increased expression of pro-inflammatory cytokines (7, 8) and/or nitric oxide (7, 8) and subsequent reduction in cytochrome function. Acute inflammation caused by interferon did not significantly altered pharmacokinetics of CEL despite a trend towards increased AUC of the drug in the inflamed as compared with control rats. Al least two explanations may be offered: 1) The enzymes responsible for metabolism of CEL are not affected by acute inflammation, and/or 2) CEL is a drug with a relatively low liver extraction efficiency, hence, its clearance is not sensitive to the inhibitory effect of inflammation.
Conclusions
The determination of CEL plasma concentrations using a newly validated liquid-liquid extraction HPLC assay has allowed us to characterize CEL PK in the rat, as well as determine CEL bioavailability. The effect of inflammation on CEL has been found to be minimal. Further extermination is required with different inflammatory models before we can conclusively determine the effects of inflammation on CEL PK.
Acknowledgements
Supported by the Canadian Arthritis Network (National Networks of Excellence) and Canadian Institute of Health Research.
Reference List
- Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents [published erratum appears in Nature 1997 Feb 6;385(6616):555]. Nature 1996;384:644-648.
- Crofford L.J. Cox-1 and Cox-2 Tissue experssion: implicatins and predictions. J Rheumatol. 1997;Suppl 49:15-19.
- Needleman P, Isakson PC. Selective Inhibition of Cyclooxygenase 2. Science and Medicine 1998;January/February 1998:26-35.
- Vane JR, Bakhle YS, Botting RM. CYCLOOXYGENASES 1 AND 2. Annu.Rev.Pharmacol.Toxicol. 1998;38:97-120.
- Geis G.S. Update on clinical developments with celecoxib, a new specific cox-2 inhibitor: what can we expect ? Journal of Rheumatology 1999;26:31-36.
- Peterson W.L., Cryer B. Cox-1-Sparing NSAIDs is the enthusiasm justified. JAMA 1999;82:1961-1963.
- Morgan E.T. Regulation of Cytochromes P450 during inflammation and infection. Drug Metabolism Reviews 1997;29:1129-1188.
- Morgan ET, Sewer MB, Iber H, Gonzalez FJ, Lee YH, Tukey RH, Okino S, Vu T, Chen YH, Sidhu JS, Omiecinski CJ. Physiological and pathophysiological regulation of cytochrome P450. Drug Metab Dispos 1998;26:1232-1240.
- Iber H., Sewer M.B., Barclay TB, Mitchell SR, Li T, Morgan E.T. Modulation of drug metabolism in infectious and inflammatory diseases. Drug Metabolism Reviews 1999;31:29-41.
- Paulson SK, Engel L, Reitz B, Bolten S, Burton EG, Maziasz TJ, Yan B, Schoenhard GL. Evidence for polymorphism in the canine metabolism of the cyclooxygenase 2 inhibitor, celecoxib. Drug Metab Dispos 1999;27:1133-1142.
- Mayo P.R., Kulmatycki K., Jamali F. Pharmacokinetic Intercation between interferon-α2a and verapamil enantiomers without pharmacodynamic consequence. Pharm Res 1996;13:S-432(Abstract)
- Paulson SK, Zhang JY, Breau AP, Hribar JD, Liu NW, Jessen SM, Lawal YM, Cogburn JN, Gresk CJ, Markos CS, Maziasz TJ, Schoenhard GL, Burton EG. Pharmacokinetics, tissue distribution, metabolism, and excretion of celecoxib in rats. Drug Metab Dispos 2000;28:514-521.
- Paulson SK, Hribar JD, Liu NW, Hajdu E, Bible RHJ, Piergies A, Karim A. Metabolism and excretion of [(14)C]celecoxib in healthy male volunteers. Drug Metab Dispos 2000;28:308-314.
- Davies NM, McLachlan AJ, Day RO, Williams KM. Clinical pharmacokinetics and pharmacodynamics of celecoxib: a selective cyclo-oxygenase-2 inhibitor. Clin Pharmacokinet 2000;38:225-242.
- Mayo P.R., Skeith K., Russell A.S., Jamali F. Decreased dromotropic responce to verapamil despite pronounced increased drug concentration in rheumatoid arthritis. Br J Clin Pharmacol 2000;50:605-13.
- Piquette-miller M., Jamali F. Selective Effect of Adjuvant Arthritis on the disposition of propranolol enantiomer in rats detected using a stereoseleective HPLC assay. Pharm Res 1993;10:294-299.
- Piquette-miller M., Jamali F. Influence of severity of inflammation on the desposition kinetics of propranolol enatiomers in ketoprofen-treated and untreated aduvant arthritis. Drug Metabolism and Disposition 1995;23:240-245.
- Emami J, Pasutto FM, Jamali F. Effect of experimental diabetes mellitus and arthritis on the pharmacokinetics of hydroxychloroquine enantiomers in rats. Pharm Res 1998;15:897-903.
Corresponding Author: Dr. F. Jamali, Faculty of Pharmacy and Pharmaceutical Sciences, University of Alberta, Edmonton, Alberta, Canada, T6G 2N8, fjamali@pharmacy.ualberta.ca
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps
CSPS Home | JPPS Home | Search | Subscribe to JPPS
|
|