J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 4(1):15-23, 2001
In Vitro Investigation of the Hepatic Extraction of RSD1070, A Novel Antiarrhythmic Compound
Vincent Tong, Frank S. Abbott1
Division of Pharmaceutical Chemistry, Faculty of Pharmaceutical Sciences, The University of British Columbia. 2146 East Mall, Vancouver, B.C., V6T 1Z3, CanadaSalome Mbofana,
Nortran Pharmaceuticals Ltd., 3650 Wesbrook Mall, Vancouver, B.C., V6S 2L2, Canada.Michael J.A. Walker
Department of Pharmacology and Therapeutics, Faculty of Medicine, The University of British Columbia, 2176 Health Sciences Mall, Vancouver, B.C., V6T 1Z3, Canada.Received December 12, 2000; Revised Febuary 2, 2001, Accepted February 3, 2001
Abstract
Purpose. The hepatic extraction of a novel antiarrhythmic, RSD1070, was investigated to test the hypothesis that the poor bioavailability observed in rats is due to high hepatic metabolism.
Methods. The pharmacokinetics of RSD1070 was examined in rats (n
=8) and its metabolism was investigated using pooled rat hepatic microsomes. The free fraction in plasma and microsomal matrices was determined by equilibrium dialysis. Hepatic extraction was predicted by scaling-up of the microsomal kinetic data using the well-stirred liver model.Results. RSD1070 demonstrated tri-exponential decay following single iv bolus administration of a dose of 12 mg/kg. RSD1070 exhibited a rapid elimination, t1/2 of 25 ± 8 min and a CLtot of 71 ± 9 mL/min/kg. Renal clearance based on 24 h urinary recovery was determined to be insignificant (<< 1% of CLtot). A Michaelis-Menten model described the elimination of RSD1070 with a Km of 0.45 mg/mL and Vmax of 2.81 mg/min/mg microsomal protein. Taking the Vmax/Km ratio (CLint) as the basis for scaling, the data from the microsomal kinetic studies (75 mL/min/kg) closely approximated the apparent CLtot. In the scale-up of the in vitro CLint, plasma free fraction (1.5%) and microsomal free fraction (15%) were determined and incorporated into the well-stirred liver model.
Conclusion. RSD1070 is a high hepatic extraction compound (E = 0.94) with a predicted CLh value that accounted for the CLtot observed in rats.
Introduction
RSD1070 [(±)-trans-[2-morpholino-1-(1-napthalene-ethyloxy cyclohexane mono-hydrochloride, Nortran Pharmaceuticals Ltd, Vancouver, BC, Canada] (figure 1) is an experimental antiarrhythmic agent currently in pre-clinical stages of development.
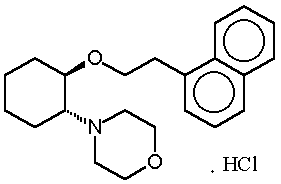
Figure 1: Chemical structure of RSD1070.
RSD1070 is an analogue of a family of compounds developed to address the need for a drug with high potency and selectivity for pathophysiologically disturbed myocardial tissue (i.e. ischemia). Current antiarrhythmic drug therapy is often unsatisfactory and has been associated with a high risk of provoking arrhythmias (1). One fundamental reason for this side effect is the poor antiarrhythmic efficacy against ischemia/infarction arrhythmias in the clinical setting (2). For example, sodium ion channel blocking agents such as encainide and flecainide lack this selectivity and are liable to be proarrhythmic at therapeutic doses and have resulted in a higher patient mortality in the randomized Cardiac Arrhythmia Suppression Trial (3).
Recently, an ester analogue of RSD1070 was developed that demonstrated increased antiarrhythmic potency under conditions of acidity and high potassium, conditions that mimic ischemia in vivo (4). RSD1070 is primarily a sodium channel blocker that shares similar "ischemia-selective" antiarrhythmic potency in rat. However, RSD1070 has demonstrated poor oral bioavailability in rats following iv and oral administration and this unacceptable bioavailability (5) has limited its ability to be considered as a clinical candidate (personal communications, Dr. R.A. Wall, University of British Columbia).
This study investigates the hepatic extraction of RSD1070 in rat using in vitro and in vivo methodologies to answer the hypothesis that the poor oral absorption observed is mainly due to high hepatic extraction. Parent compound disappearance in microsomal metabolism studies for the calculation of intrinsic clearance is described. Resultant intrinsic clearances were "scaled-up" to reflect the whole liver clearance using a published strategy (6). The free fraction of RSD1070 in rat plasma and in microsomal incubates was determined for the prediction of hepatic extraction using the well-stirred liver model.
Methods and materials
Chemicals. RSD1070 (MW 376, pKa 7.8) and RSD921 (internal standard, MW 393, pKa 9.3) were supplied by Nortran Pharmaceuticals Ltd. (Vancouver, BC, Canada). RSD1070 purity was greater than 99% by GC-FID, and the internal standard was greater than 99% by NMR. Methyl tert-butyl ether (HPLC grade) was purchased from Fischer Scientific (Vancouver, BC, Canada). Methanol (HPLC grade) was obtained from Caledon Laboratories Ltd. (Georgetown, ON, Canada). NADPH tetrasodium salt was from Boehringer Mannheim (GMBH, Germany).
Animals and treatment. Male Sprague-Dawley rats (200 300 g) obtained from the University of British Columbia Animal Care Facility, were housed two per cage on a bedding of corn-cob and fed with rat diet and water ad libitum. Animals were maintained in a holding room on a 12 h light/12 h dark cycle at constant temperature (22°C) and humidity.
Pharmacokinetic studies. All animal experimentation was approved by the University of British Columbia Animal Care Committee (Vancouver, BC, Canada). Rats were cannulated with PE-20 tubing at the abdominal aorta and the inferior vena cava under anesthesia (sodium pentobarbital, 65 mg/kg, ip). The cannulae were passed through a trocar and exteriorized by threading the trocar under the skin of the back and out through a small incision at the mid-scapular region. All incision sites were closed with silk sutures and the animal was housed individually in a cage with food and water ad libitum. At least 24 h was allowed for recovery before commencing with the experiment. Rats were administered a single iv bolus antiarrhythmic ED90 dose of 12 mg/kg via the inferior vena cava cannula and housed in stainless steel metabolic cages equipped with a glass bottle to collect urine. All animals were allowed access to food and water ad libitum. Blood samples (250
mL) were collected at 2.5, 5, 10, 15, 20, 30, 45, 60, 120, 240, 360, and 540 min post administration in Eppendorf microcentrifuge tubes via the inferior vena cava cannula using heparinized syringes. All blood samples were immediately centrifuged (13,500 x g for 5 min at room temperature) and the plasma removed and stored at 20° C until further analysis. Twenty-four h urine was collected in metabolic cages and also frozen at 20° C until further analysis.Preparation of pooled hepatic microsomes. Rats (n
=4) were sacrificed by decapitation, the liver was immediately removed, weighed, and placed into homogenizer tubes on ice with ice-cold 0.05 M Tris-HCl/ 1.15% KCl buffer. Livers were homogenized, the homogenate pooled into centrifuge bottles and centrifuged at 9,000 x g for 20 min at 4° C. The supernatant (S-9) was filtered through gauze and centrifuged at 105,000 x g for 60 min at 4° C. The resulting microsomal pellet was resuspended in 30 mL of 0.25 M sucrose solution. Aliquots of microsome preparation (0.5 mL) were stored frozen in Cryovials (Ingram & Bell, Richmond, BC, Canada) at 70° C. Total cytochrome P450 content was assayed by the method of Omura and Sato (7). Microsome protein was determined by the method of Lowry et al. (8).Hepatic microsomal studies. The disappearance time-profile for RSD1070 in microsomal incubations was monitored. Incubates (950
mL) contained pooled rat liver microsomal protein (0.1 mg/mL) and 1.5 mM NADPH in 50 mM KH2PO4/K2HPO4 buffer (pH 7.4) with 3 mM MgCl2. The mixture was gently mixed, preincubated for 5 min at 37° C, and the reaction was initiated with the addition of 50 mL RSD1070 (substrate). Final substrate concentrations were 0.34, 0.85, 1.7, 3.4, and 8.5 mg/mL (1 25 mM). Reactions were terminated with the addition of cold 2M NaOH (100 mL) after 0, 2.5, 5, 10, 15, 20, and 30 min of incubation and vortex-mixed. The samples were placed on ice prior to analysis. All incubations were conducted in duplicate.Protein binding studies and determination of blood to plasma partition coefficient. Protein binding was determined by equilibrium dialysis. Triplicate samples were prepared by spiking known amounts of RSD1070 in blank rat plasma for final concentrations of 1, 5, and 10
mg/mL and followed with incubation in a shaking water bath (2 h at 37° C). Samples were dialyzed against isotonic phosphate buffer (pH 7.4) in plexiglass dialysis cells (1 mL capacity). Membrane dialysis sacs (Sigma Diagnostics, Inc., St. Louis, MO) with a molecular weight cutoff of 12,000 MW were utilized. Prior to use, the membranes were conditioned by boiling for 30 min in distilled water and for another 30 min in dialysis buffer. Equal volumes of plasma sample and phosphate buffer (0.8 mL) were transferred to opposite sides of the membrane. Equilibrium dialysis was performed at 37° C for 5 h in a shaking water bath. The time to reach equilibration was defined as the time required for a plot of drug concentration in plasma buffer against time to asymptote. The recovery and stability of RSD1070 from both sample and plasma reservoirs were assessed and were found to be highly recovered (> 90% of total drug) and stable (< 10% deviation) following 6 h of incubation at 37° C.The free fraction in microsomal matrix was determined by equilibrium dialysis. RSD1070 (1.7
mg/mL) was incubated with 0.1 mg/mL microsomal protein in microsomal incubation buffer for 30 minutes at 37° C. NADPH cofactor was excluded to prevent turnover of RSD1070. The sample was transferred to equilibrium dialysis units and dialyzed against incubation buffer (0.8 mL each) for 5 h at 37° C. The fractions unbound were calculated from the ratio of unbound concentration in buffer reservoir to the total concentration in sample reservoir.The blood to plasma partition concentration ratio was calculated from the ratio of RSD1070 concentration determined in whole blood and plasma after incubating RSD1070 (10
mg/mL) in whole blood for 2 h at 37°C.Assay procedure for the determination of RSD1070 concentrations. All samples were analyzed by an LC/ MS/MS assay previously described and validated by Tong et al., (manuscript in progress). Samples, standards, and quality control samples were processed by a single step, liquid-liquid base extraction (pH 13). Internal standard (100
mL, 0.5 mg/mL) and 2M NaOH (100 mL) were added to the sample and vortex-mixed. Methyl tert-butyl ether (5 mL) was added to each sample and vortex-mixed briefly prior to and after placement in a LabquakeÒ mixer (LabIndustries, Berkeley, CA, USA) for 30 min. The separation of the aqueous and organic layers was aided by centrifugation (2000 x g for 10 min). The organic layer was dried under a gentle stream of nitrogen (20 min at 30°C, 0.5 PSI). Samples were reconstituted in 1 mL of mobile phase (40% MeOH, 0.2% formic acid).LC/MS/MS detection and quantitation of RSD1070 was carried out using a Fisons VG Quattro (Altrinchem, UK) tandem mass spectrometer interfaced with a Hewlett Packard (Avondale, PA) 1090 II Liquid chromatograph. Positive electrospray was used as the means of ionization and collision induced dissociation (CID) involved argon as the target gas pressure of » 3.5 x 10
-3 mBar. Other parameters were capillary voltage 3 kV, cone voltage 30 V with skimmer offset by 5 V, and collision energy 40 eV. The multipliers 1 and 2 were both set at 650 V. The low mass and high mass resolutions were set at 12.5/12.5 for MS1 and 5.0/5.0 for MS2. The source temperature was 140°C. MS/MS experiments were carried out on protonated molecular ions. Mass selective detection of RSD1070 and IS were performed by multiple reaction monitoring (MRM) with a dwell time of 0.3 seconds/channel and with an inter channel delay of 0.03 seconds. Parent-daughter ion transitions detected were m/z 340 > 155 (RSD1070) and m/z 357 > 147 (IS). RSD1070 concentrations were determined from peak area ratios of RSD1070 to IS obtained from LC/MS/MS chromatograms.The HPLC was fitted with a Phenomenex Columbus C18 (150 mm x 2 mm, 5
m) column (Torrance, CA, USA), and samples (10 mL) were delivered at a flow rate of 0.2 mL/min. The mobile phase consisted of methanol/water (0.2% FA) and was programmed: linear gradient from 40 to 80% methanol during 0 to 8 minutes, a gradient decrease to 40% methanol during 8 to 8.5 min, and a hold at 40% methanol to 12 minutes.Assay validation was previously performed in both rat plasma and rat liver microsomal matrices. The extraction recovery of RSD1070 was greater than 95% and the assay was linear over the concentration range of 2.5-100 ng/mL. Inter-assay and intra-assay variability (coefficient of variation) was approximately 20% at the limit of quantitation (2.5-3 ng/mL) and less than 15% at all other concentrations. The accuracy of the assay (% bias of nominal concentration) was less than 15%.
Data analysis
In vivo calculations. The pharmacokinetic parameters of the RSD1070 plasma concentration-time curve after iv administration in rat were derived using non linear regression analysis and single iv bolus non-compartment modeling (WinNonlin, v1.1, Scientific Consulting Inc., Apex, NC, USA). The area under the plasma concentration-time curve (AUC¥ ) was calculated by trapezoidal rule with extrapolation to infinity. The elimination rate constant,
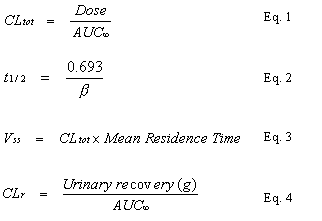
b, was calculated from the slope of the terminal elimination phase. The apparent pharmacokinetic parameters, total body clearance (CLtot), elimination half-life (t1/2), volume of distribution at steady state (Vss), and renal clearance (CLr) were calculated by the following equations (Eq. 1-4):
In vitro calculations. Kinetic analysis of the in vitro consumption of RSD1070 was undertaken using nonlinear regression (SigmaPlot, v.5.0). The plot of initial reaction rate (
no) against substrate concentration (S) was hyperbolic and was found to be satisfactorily described by a standard Michaelis-Menten equation (Eq. 5):

Where
no and Vmax are the observed and maximal rates of metabolism, respectively and Km is the Michaelis constant. The enzyme kinetic parameters were also determined by an Eadie-Hofstee plot of the microsome metabolism data.In vitro intrinsic clearance (CL
int) was calculated from the ratio of the enzyme kinetic parameters under linear conditions such that [S] << Km (Eq. 6):

Where V is the microsomal incubation volume.
In vitro CL
int was scaled to an in vivo CLint¢ (in units of mL/min/kg) using experimentally determined microsomal protein yield (mg/g of liver) and average liver weight (g of liver/ kg body weight) according to the following formula (6) (Eq. 7):
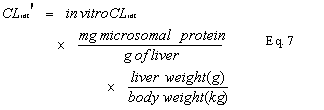
Using the well-stirred (venous equilibrium) liver model (9,10) with consideration to non-specific binding to microsomal protein (11), the hepatic plasma clearance (CLh), can be described by the following relationship (Eq. 8):
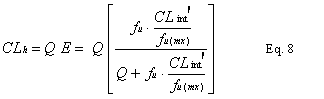
Where Q is hepatic blood flow having a literature value of 80 mL/min/kg (12), f
u the free fraction of drug in plasma, fu(mx) the free fraction of drug in microsomal matrix, and hepatic intrinsic clearance (CLint), and E is the hepatic extraction ratio.Results
In vivo experiments
The time course of mean plasma concentrations of RSD1070 following a single iv bolus dose of 12 mg/kg (n=8) is illustrated in figure 2.
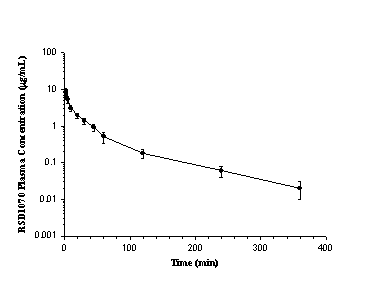
Figure 2: Mean concentration-time plot of RSD1070 in plasma following a single iv bolus dose of 12 mg/kg of RSD1070 in rats. Each value represents the mean concentration obtained from 8 animals at each time point. Error bars represent the SD.
A summary of the pharmacokinetic parameters is shown in table 1.
Table 1: Calculated pharmacokinetic parameters of RSD1070 for individual animals based on rat plasma and urine concentrations following single iv bolus administration of a dose of 12 mg/kg (n=8).

After iv administration the plasma concentration-time curve exhibited tri-exponential decay with a rapid elimination t
1/2 of 25 ± 8 min, a Vss of 2.8 ± 0.4 L/kg, and CLtot of 71 ± 9 mL/min. Twenty-four h urine concentrations accounted for << 1% of the dose and resulted in an insignificant renal clearance (<< 1% of CLtot).In vitro experiments
The total metabolism of RSD1070 was monitored as a function of time in pooled rat liver microsome saturation kinetic studies. The parent compound disappearance profile of RSD1070 at substrate concentrations of 0.34 to 8.5
mg/mL (1 25 mM) is shown in figure 3.
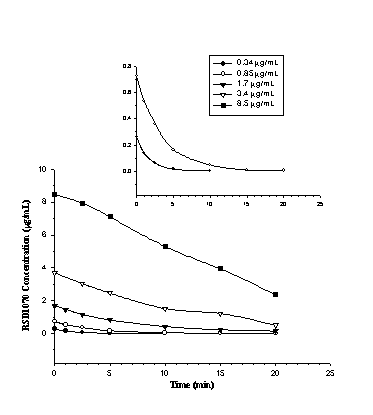
Figure 3: RSD1070 concentration versus time profiles in rat liver microsomal incubations. Initial concentrations of RSD1070 ranging from 0.34 8.5 mg/mL were incubated with 0.1 mg/mL of pooled rat liver microsomes and 1.5 mM NADPH. Reaction was terminated at various time points with the addition of 2M NaOH. All samples were processed and analyzed as described in the text. The insert diagram shows the disappearance profile for the lower substrate concentrations of 0.34 and 0.85 mg/ mL on a smaller y-axis scale.
RSD1070 was rapidly eliminated in rat liver microsome incubations. Consumption rates (
no) of RSD1070 was calculated from the slope of the initial linear decline at each respective substrate concentration and ranged from 1.2 to 2.7 mg/min/mg protein (table 2).Table 2: Initial rate (no) of RSD1070 metabolism catalyzed by microsomal enzymes for each starting substrate concentration. Incubation conditions consisted of 0.1 mg/mL microsomal protein and 1.5 mM NADPH. The initial rates were calculated as the negative slope of the initial linear decline from the parent compound disappearance profile in figure 3.
The plot of initial reaction rate (
mg/min/mg protein) against substrate concentration (mg/mL) was hyperbolic and could be described by classical Michaelis-Menten kinetics with a Vmax of 2.81 mg/min/ mg protein and Km of 0.45 mg/mL (figure 4).
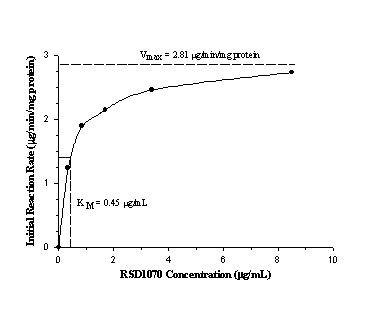
Figure 4: Relationship between initial linear rate of RSD1070 disappearance and starting concentrations of RSD1070 in a typical microsomal incubation. Vmax and Km were estimated by model fitting the data to the standard Michaelis-Menten equation using the Sigma Plot (v5.0) program.
The Eadie-Hofstee plot of the microsomal disappearance rates of RSD1070 versus the initial RSD1070 concentrations is shown in figure 5.
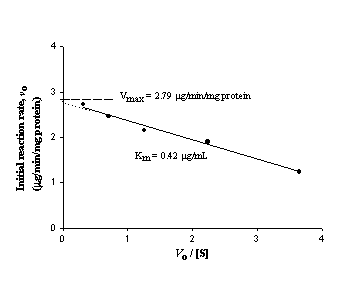
Figure 5: Eadie-Hofstee plot for the determination of Vmax and Km describing the consumption of RSD1070 in microsomal incubations. The relationship between the initial reaction rate noversus no/[S] is plotted, and a straight line is obtained where the slope is equal to Km and the y-intercept is equal to Vmax.
The calculated V
max (2.79 mg/min/mg protein) and Km (0.42 mg/mL) were in close approximation to the values obtained by model fitting the data to the standard Michaelis-Menten equation. The in vitro CLint calculated from the ratio of the enzyme kinetic parameters (Vmax/Km) was estimated to be 6.2 mL/min/mg protein (Eq. 6).RSD1070 plasma protein binding was determined in vitro over the concentration range of 2-20
mg/mL with a mean unbound fraction of 1.5 ± 0.5% (n=3). The mean free fraction in microsomal incubates was determined to be 15 ± 2% (n=3) over the concentration range of 1.7 - 10 mg/mL of RSD1070. The blood to plasma concentration ratio for RSD1070 was close to unity, 0.95 ± 0.05 (n=3).In vitro prediction of hepatic extraction
The in vivo CL
int, reflective of RSD1070 metabolism in the whole liver, was obtained by multiplying the in vitro CLint (mg/min/mg microsomal protein) by the microsomal protein content (per gram liver) and by the liver weight (per kg of rat body weight) as described in Eq. 7. Microsomal protein content was experimentally determined to be 40 mg protein/ g of liver and the average rat liver weight was 47 g/ kg body weight. In vivo CLint was estimated to be 1.2 x 104 mL/min/kg. There was close approximation between the predicted hepatic clearance (75 mL/min/kg) and the apparent total body clearance (71 mL/min/kg), which suggests that RSD1070 is a high clearance compound (» hepatic blood flow) that may be primarily eliminated by liver metabolism. Thus, RSD1070 is considered to be highly extracted by the liver with a predicted extraction ratio value of 0.94. The results are summarized in table 3.Table 3: Predicted hepatic clearance and hepatic extraction ratio values from microsomal intrinsic clearance values analyzed by enzyme kinetic method.
Discussion
The prediction of hepatic extraction for novel pharmaceutical compounds has important ramifications in the drug discovery process. The application of in vitro studies to identify and exclude potential new chemical entities that would be expected to exhibit unsatisfactory human pharmacokinetics (e.g. poor bioavailability) prior to clinical trials has been recently reviewed (13,14,15). Central to the prediction of in vivo drug metabolism from in vitro metabolism data through the use of liver S9 fraction, microsomes, and hepatocytes is the calculation of CL
int, and several methods and approaches have been described (6). The concept of in vitro-in vivo correlation was first examined by Rane et al. (16) with the use of hepatic (S9) fractions to estimate the extent of hepatic extraction from the Michaelis-Menten parameters (Vmax and Km) of several compounds. This traditional enzyme kinetic approach measures CLint from the Michaelis-Menten parameters (Vmax and Km) for the formation of the major metabolite(s). This approach is feasible when measuring formation rates for compounds that are primarily metabolized to major metabolites without subsequent metabolism as in the case of phenytoin and tolbutamide (17), or when measuring formation rates for total metabolite production as in the case of caffeine (18). As an alternative approach, Obach (19) demonstrated the applicability of calculating the in vitro t1/2 from the 1st order elimination rate constant under linear microsomal conditions for the prediction of human clearance.Our study utilized pooled rat hepatic microsomes to test the hypothesis that extensive hepatic extraction contributes to the poor oral bioavailability of RSD1070 observed in rats. For this purpose, the formation rate of the N-dealkyl metabolite of RSD1070 was initially investigated in microsomal incubations; however, sequential metabolism of this metabolite has made this improbable. Therefore, this study examined the parent compound lability in hepatic microsomal metabolism studies for the determination of the overall consumption rate. The use of drug disappearance time profiles to assess metabolic clearances from kinetic parameters has been demonstrated with compounds such as felodipine (20) and diazepam (21). Hybrid Michaelis-Menten parameters, V
max and Km, describing the sum of all pathways contributing to the consumption of RSD1070 were estimated from the enzyme kinetic saturation plot of initial reaction rate and substrate concentration. A simple one-site model described the data with an approximate hybrid Km of 0.45 mg/mL and a Vmax of 2.81 mg/min/mg protein. Thus under linear conditions ([S] << Km, substrate concentrations below 45 ng/mL), the predicted hepatic clearance was estimated to be 75 mL/min/kg based on the well stirred-liver model. This estimate was obtained by the ratio of Vmax and Km, which were also estimates based on limited data points. In particular, the apparent Km was an approximation because it was based on one data point that was below the apparent Km value. However, further analysis of the kinetic data using an Eadie-Hofstee plot resulted in similar estimates of Vmax and Km values. The estimate of the hybrid Km would be further strengthened with additional data points below the apparent Km value. The attempt to further delineate the disappearance profile with substrate concentrations below 0.34 mg/mL (1 mM) was not successful because of the very high turnover rate and the limits of the assay.The free fraction of RSD1070 in plasma as well as in the microsomal incubation medium was considered for the calculation of hepatic extraction according to Eq. 8. Free fractions of drug in microsomal incubation media were used for the correction of K
m in earlier in vitro-in vivo correlation studies with felodipine (20) ethoxybenzamide (22,23), imipramine and desipramine (24), and diazepam and analogues (25). The importance of non-specific binding to microsomal incubation matrix has been investigated (11,19,26) and the studies demonstrated that the inclusion of both free fraction parameters in plasma and microsomes resulted in the best agreement between in vivo clearance values and clearance values projected from in vitro CLint data. The extensive protein binding of RSD1070 (a basic compound with a pKa of approximately 8) is consistent with the overall trend that basic lipophilic amine compounds demonstrate extensive binding to plasma proteins and to microsomal proteins compared to neutral and acidic compounds (19).The lack of information regarding the disposition and the metabolic profile of RSD1070 prompted a preliminary pharmacokinetic study in rats. RSD1070 appeared to be highly eliminated in rat with a rapid elimination half-life and a high total body clearance value that approximated the literature value rat hepatic blood flow of 80 mL/min/kg (12) and the predicted hepatic clearance. Because the total body clearance is comprised of the sum of individual organ clearances, the in vivo studies do not provide any direct information regarding the extent of hepatic extraction. More specific in vivo studies requiring drug administration via the hepatic portal vein is required for a direct evaluation of hepatic extraction.
Based on the hepatic microsomal studies, our results are consistent with the hypothesis that RSD1070 is a high hepatic extraction compound. The microsomal metabolic clearance of the liver is involved in the elimination of RSD1070 to a large extent with a high predicted hepatic clearance approximating the observed total body clearance. Further studies are required to investigate the contribution of intestinal absorption and metabolism as extra-hepatic factors that may reduce the overall bioavailability of RSD1070. With regards to the prediction of human bioavailability, further metabolism studies using human microsomes or hepatocytes may also be investigated.
Abbreviations
CL
int, intrinsic clearance based on in vitro dataCL
int¢ , in vivo intrinsic clearance scaled-up from in vitro dataCL
h, hepatic clearanceCL
tot, total body clearanceE, hepatic extraction ratio
f
u, fraction unbound in plasmaf
u (mx), fraction unbound in microsomal matrixIS, internal standard
K
m, Michaelis constantQ, hepatic blood flow
V
max, maximal velocityAcknowledgements
The authors thank Nortran Pharmaceuticals Ltd. for the provision of RSD1070 and internal standard, Dr. Lillian Clohs and Mr. Mark Buss (Nortran Pharmaceuticals Ltd.) for discussion in analytical method development, and Mr. Harvey Wong (Faculty of Pharmaceutical Sciences, UBC) for assistance in the pharmacokinetic studies. Technical assistance from Mr. Roland Burton (Faculty of Pharmaceutical Sciences, UBC) in the use of LC/MS/MS is greatly appreciated. Funding to Vincent Tong from Nortran Pharmaceuticals Ltd. is acknowledged.
References
Roden, D.M., Risks and benefits of antiarrhythmic therapy. N Engl J Med, 331:785-791, 1994.
Echt, D.S., Liebson, P.R., Mitchell, L.B., Peters, R.W., Obias-Manno, D., Barker, A.H.,D. Arensberg, D.,Baker, A., Friedman, L., Greene, H.L., Huther, M.L., Richardson, D.W., and the CAST Investigators, Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The cardiac arrhythmia suppression trial. N Engl J Med, 324:781-788, 1991.
CAST Investigators, Preliminary report: effect on encainide and flecainide on mortality in a randomized trial of arrhythmia suppresion after myocardial infarction. N Engl J Med, 321:406-412, 1989.
Yong, S.L., Xu, R., Mclarnon, G., Zolotoy, A.B., Beatch, G.N., and Walker, M.J.A., RSD1000: a novel antiarrhythmic agent with increased potency under acidic and high-potassium conditions. J Pharmacol Exp Ther, 289:236-244, 1999.
Prentis, R.A., Lis, Y., and Walker. S.R., Pharmaceutical innovation by the seven UK-owned pharmaceutical companies (1964-1985). Br J Clin Pharmacol, 25:387-396, 1988.
Houston, J.B., Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol, 47:1469-1479, 1994.
Omura, T. and Sato, R., The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J Biol Chem, 239:2379-2385, 1964.
Lowry, O.H., Rosebrough, N.J., Farr, A.L., and Randall, R.J., Protein measurement with the Folin phenol reagent. J Biol Chem, 193:265-275, 1951.
Rowland, M., Benet, L.Z., and Graham, G.G., Clearance concepts in pharmacokinetics. J Pharmacokinet Biopharm, 1:123-136, 1973.
Wilkinson, G.R. and Shand, D.G., A physiological approach to hepatic drug clearance. Clin Pharmacol Ther, 18:377-390, 1975.
Obach, R.S., The importance of non-specific binding in in vitro matrices, its impact on enzyme kinetic studies on drug metabolism reactions for in vitro-in vivo correlations. Drug Metab Dispos, 24:1047-1049, 1996.
Pollack, G.M., Brouwer, K.L.R., Demby, K.B., and Jones, J.A., Determination of hepatic blood flow in the rat using sequential infusions of indocyanine green and galactose. Drug Metab Dispos, 18:197-202, 1990.
Iwatsubo, T., Hirota, N., Ooie, T., Suzuki, H., Shimada, N., Chiba, K., Ishizaki, T., Green, C.E., Tyson, C.A., and Sugiyama, Y., Prediction of in vivo drug metabolism in the human liver from in vitro metabolism data. Pharmacol Ther, 73:147-171, 1997.
Obach, R.S., Baxter, J.G., Liston, T.E., Silber, B.M., Jones, B.C., MacIntyre, F., Rance, D.J., and Wastall, P., The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther, 283:46-58, 1997.
Lavé, T., Dupin, S., Schmitt, C., Valles, B., Ubeaud, G., Chou, R.C., Jaeck, D., and Coassolo, P., The use of human hepatocytes to select compounds based on their expected hepatic extraction ratios in humans. Pharm Res, 14:152-155, 1997.
Rane A., Wilkinson, G.R., and Shand, D.G., Prediction of hepatic extraction ratio from in vitro measurement of intrinsic clearance. J Pharmacol Exp Ther, 200:420-424, 1977.
Ashworth, E.I.L., Carlile, D.J., Chenery, R., and Houston, J.B., Prediction of in vivo disposition from in vitro systems: clearance of phenytoin and tolbutamide using rat hepatic microsomal and hepatocyte data. J Pharmacol Exp Ther, 274:761-766, 1995.
Hayes, K.A., Brennan, B., Chenery, R., and Houston, J.B., In vivo disposition of caffeine predicted from hepatic microsomal and hepatocyte data. Drug Metab Dispos, 23:349-353, 1995.
Obach, R.S., Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and non-specific binding to microsomes. Drug Metab Dispos, 27:1350-1359, 1999.
Bäärnhielm, C., Dahlbäck, H., and Skänberg, I., In vivo pharmacokinetics of felodipine predicted from in vitro studies in rat, dog, and man. Acta Pharmacol Toxicol, 59:113-122, 1986.
Igari, Y., Sugiyama, Y., Sawada, Y., Iga, T., and Hanano, M., In vitro and in vivo assessment of hepatic and extrahepatic metabolism of diazepam in the rat. J Pharm Sci, 73:826-828, 1984.
Lin, J.H., Hayashi, M., Awazu, S., and Hanano, M., Correlation between in vitro and in vivo drug metabolism rate: oxidation of ethoxybenzamide in rat. J Pharmacol Exp Ther, 6:327-337, 1978.
Lin, J.H., Sugiyama, Y., Awazu, S., and Hanano, M., Kinetic studies on the deethylation of ethoxybenzamide. Biochem Pharmacol, 29:2825-2830, 1980.
Chiba, M., Fujita, S., and Suzuki, T., Pharmacokinetic correlation between in vitro hepatic microsomal enzyme kinetics and in vivo metabolism of imipramine and desipramine in rats. J Pharm Sci, 79:281-287, 1990.
St. Pierre, M.V. and Pang, K.S., Concentration-dependent metabolism of diazepam in mouse liver. J Pharmacokinet Biopharm, 23:243-266, 1995.
Obach, R.S., Nonspecific binding to microsomes: impact on scale-up of in vitro intrinsic clearance to hepatic clearance as assessed through examination of warfarin, imipramine, and propranolol. Drug Metab Dispos, 25:1359-1369, 1997.
Corresponding Author: Frank S. Abbott, Faculty of Pharmaceutical Sciences, UBC, 2146 East Mall, Vancouver, British Columbia, Canada, fabbott@interchange.ubc.ca
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps



