J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 3(3):281-291, 2000
Significance Of Apoptosis And Its Relationship To Antioxidants After Ochratoxin A Administration In Mice
Faik Atroshi1, Isa Biese, Hannu Saloniemi
Department of Clinical Sciences, Pharmacology & Toxicology, Faculty of Veterinary Medicine, Helsinki University, FinlandTerhi Ali-Vehmas, Seppo Saari
Department of Basic Veterinary Sciences, Faculty of Veterinary Medicine, Helsinki University, FinlandAldo Rizzo, Pirjo Veijalainen
Department of Virology & Epidemiology, National Veterinary and Food Research Institute, Helsinki, FinlandManuscript received September 18, 2000, Revised November 2, 2000, Accepted November 3, 2000
PDF version for printing
ABSTRACT
A study of the appearance of liver apoptosis after ochratoxin A (OTA) administration was performed in male mice. Administration of OTA twice a week for one or two weeks period results in the occurrence of apoptosis in mice's liver. The presence of intracellular apoptosis bodies was detected at two weeks after toxin treatment. Light microscopic examination demonstrated the presence of eosinophilic globules, often containing apoptotic bodies. They were found within the cytoplasm of intact hepatic cells. The number of apoptotic bodies was further enhanced at two weeks, resulting in 8 fold increases in liver over the control values. No evidence of cell necrosis could be observed by histological and biochemical analysis at one week. However, centrilobular necrosis was evident at two weeks. The ability of the combined antioxidants: Coenzyme Q 10 (CoQ 10), L-carnitine, Zn, Mg, N-acetyl cysteine, vitamin C, vitamin E and selenium or tamoxifen to intervene in apoptosis induced in livers of mice by OTA was also investigated. The inhibition by these scavengers was more clear in mice treated with OTA for one week than those mice treated for two weeks. Treatment with tamoxifen, known in restoration of tumor suppressor function and on induction of programmed cell death (apoptosis), after OTA administration, had no significant inhibition effect on the incidence of apoptotic bodies in liver. Because hepatic glutathione represents the major defence against toxic liver injury, we studied the activity of tissue reduced glutathione (GSH), known to inhibit apoptosis. Our finding showed that two weeks after treatment, OTA caused a decrease of the GSH activity. However, treatment of mice with the combined antioxidants could enhance hepatic antioxidant/detoxification system, as indicated by increase in hepatic reduced glutathione level. In the light of these results, apoptosis was observed after two weeks of OTA administration. We also suggest that use of the combined antioxidants may be of interest in conditions were certain toxin-mediated forms of cell death and/or apoptosis contribute significantly to toxicity.
INTRODUCTION
Ochratoxin A (OTA) is a nephrotoxic, hepatotoxic and teratogenic mycotoxin produced by storage molds (mainly by species of Aspergillus and Penicillium ) on a variety of commodities. Exposure to low concentrations of this toxin causes morphological and functional changes in kidney and liver of several domestic and experimental animals. The toxin has also been found in human sera from people living in areas where Balkan endemic nephropathy occurs, and it is suggested to be a possible determinant of this fatal human disease (1). The molecular mechanisms involved in OTA-induced nephrotoxicity (2), carcinogenesis (3, 4), teratogenic effects (5), immunosupression (6, 7), and inhibition of mitosis (8) are not clearly defined. The genotoxic effects, inhibition of DNA synthesis and mitosis, as well as histopathological effects on the nuclei of OTA-treated cells (5) may be explained by OTA-inflicted DNA damage, which include DNA adduct formation and DNA single strand breaks. OTA-DNA adducts in intoxicated mice kidney tissue has been reported (9). However, the degree of OTA-induced DNA adduct formation is dose and time dependent. Moreover, OTA-DNA adducts in a biopsy of kidney tissue from a patient suffering from chronic interstitial nephropathy and having high OTA concentration in blood has been found (10). Evidence that OTA-induced renal tubular lesions were in fact partially due to apoptosis has been demonstrated (11).
Chronic exposure to low doses of OTA in vivo caused renal adenomas and hepatocellular carcinomas in mice and rats (3, 12). Low doses of OTA in vitro caused chromosome aberrations in cultured human lymphocytes (13) and in vivo in mice (14). In acute toxicity studies, OTA-induced cell death was reported in vivo in rat renal tubules (15) and in developing mouse embryos (5), and in vitro in human lymphocytes (11,16) and in HL-60 human promyelotic leukemia cells (17). The type of cell death observed in the nephrotoxicity (15) and teratogenic studies (5) remain unclear. Cell death can generally proceed via necrosis or apoptosis [programmed cell death (PCD)] (18,19). Necrosis is characterized by the formation of tubular lesions (pores) in the plasma membrane. Apoptosis causes cell death in a way that differs morphologically and biochemically from necrosis. Earlier studies showed that the common core mechanism of apoptosis is a DNA fragmentation and morphological lesion, such as condensation and fragmentation of the nucleus and cytoplasm (20). It is now becoming apparent, however, that morphological characteristics of apoptosis are not always associated with the ladder-type DNA fragmentation and that it is probably an epiphenomenon (21,22). From studies on a variety of organisms and cell lines, many distinguishing features of apoptosis have been discovered. It is interesting that membrane sphingolipids are now believed to play a role in the transduction of the apoptoic signal (23). Cell death in host response to mycotoxins has not been fully characterized.
Apoptosis can be triggered in two principal ways: by toxic chemicals or injury leading to damage of DNA or of other important cellular targets, and activation or inactivation of receptors by growth-regulating signal factors in the organism (24). Initiation of apoptosis can result from multiple stimuli, including heat, toxins, free radicals, growth factor withdrawal, cytokines such as transforming growth factor-beta, loss of matrix attachment, glucocorticoid, nitric oxide, and radiation (25,26). These stimuli work in conjunction with other intrinsic factors that determine the cell's potential to undergo apoptosis (27). Cell death occurs normally in the liver but this has also been associated with hepatic disorders such as bile-salt-induced apoptosis (28). The recent demonstration that hepatic organic anion transporting protein mediate Na+-independent transport of a wide range of amphipathic substrates among them numerous xenobiotics (e.g, ochratoxin A) (29).
Oxidative damage may be one of the manifestations of cellular damage in the toxicity of ochratoxin A. Reactive oxygen species (ROS) have a major role in the mediation of cell damage. Several defence mechanisms attempt to minimize the production and the action of harmful oxidants. Such defence mechanisms include enzymes such as superoxide dismutase, catalase, and glutathione peroxidase, as well as natural lipophilic and hydrophilic antioxidants. Free radical damage initially induced by mycotoxins can be propagated and magnified by lipid peroxidation chain reactions (30). The notion that ROS participates in PCD regulation based on I) excessive ROS or inhibitions of antioxidant pathways induce apoptosis (31,32). Leakage of high-energy electrons along the mitochondrial electron transport chain, which causes the formation of superoxide anion radicals, has been speculated to be involved in neurodegenerative disorders that are triggered by toxins, hereditary mitochondrial DNA (mtDNA) mutation and acquired mtDNA alterations accumulating with age (33). II) When stimulated to undergo PCD, cells hyper produce superoxide anion, as determined with the fluorochrome dihydroethidine (34). III). In some cases, antioxidants and free radical scavengers inhibit the induction of PCD. The antioxidant vitamin E and C, as well as exogenous catalase, prevent PCD in some systems (35). Similarly, transfections with the selenoenzyme, glutathione peroxidase (GSH-Px), inhibits apoptotic cell death after growth factor deprivation of an IL-3-dependent cell line (31).
Tamoxifen is a membrane antioxidant in that it protects model and cellular membranes, including the nuclear membrane, fungal infection, against the potentially carcinogenic free radical intermediates and the products of lipid peroxidation (36). However, some studies have shown the potential relevance of the oxidation products of 4-hydroxytamoxifen (4-OHTAM) in carcinogenesis. Other studies show 4-OHTAM has antioxidant properties (37). Current concerns about tamoxifen are the development of rat liver tumors during long-term treatment and increased incidence of endometrial carcinomas observed in patients. Another concern is the development of drug resistance to long-term tamoxifen therapy (38).
Ochratoxin A is a nephrotoxin of environmental significance. Its characteristics nephrotoxic effects have been observed in humans (Balkan Endemic Nephropathy) as well as in a wide variety of other species e.g. in mice. In all of these species the main target organ of OTA is the kidney. Herein, we demonstrate that the liver of mice treated with OTA have also morphologic and biochemical features characteristic of apoptosis. Furthermore, the effect of membrane damage caused by OTA was examined using combined free-radical scavengers namely: Coenzyme Q 10, L-carnitine, Zn, Mg, N-acetylcystein, vitamin C, vitamin E and selenium. Further more, the effect of treatment with tamoxifen, a membrane antioxidant, on OTA-induced apoptosis is also described. The levels of GSH, a presumptive marker for mycotoxins toxicity, was investigated.
MATERIALS AND METHODS
Animals and diet. A total of 100 male inbred Han:NMRI mice 3 months old and weighing approximately 29 g, were used in this study. Based on previous investigation (39), 70 mice were randomly allocated into seven groups of ten animals. The animals were given lab chow and tap water ad libitum in an environment of controlled temperature, humidity, and light/dark cycle for 1 week prior to study. Water was freely available throughout the course of the experiment. Weight and food intake were analysed at regular intervals during the treatment. The animals, except one normal control group, were fed for 10 weeks a constant semi-synthetic feed (40), deficient in vitamin C, vitamin E and selenium used in this study. The feed contained 10% of a mixture of soya oil and lard (1:1), 2% cellulose powder, 26% maize starch, 20% casein, and 6% of a mixture of mineral salts. Mean values for consumed diet evaluated every day during the first week and 2 to 3 times per week for the rest of the treatment period. Two mice were lost before baseline measurements could be collected. The following groups of animals were used in the experiment: 1) control group or control diet not supplemented with antioxidants or OTA, 2) control deficient group, 3) combined antioxidants group, 4) tamoxifen group, 5) OTA group, 6) OTA plus combined antioxidants group and 7) OTA plus tamoxifen groups. At designated intervals mice were killed. Additional experiments also were performed wherein 20 mice given the same doses of OTA were killed at different days after treatment together with their respective controls. All animal experiments were performed in accordance with institutional guidelines. Groups 1 and 2 were killed at day 0. Livers were collected, and the hepatic tissue samples were excised and rinsed with ice-cold homogenising buffer (50 mM Tris, 0.1 mM EDTA, pH 7.6). The rest of the tissue samples were frozen in liquid N2 and stored at -85°C.
Pretreatment of mice with combined antioxidants and tamoxifen. The animals (10 mice/group) were pretreated for 10 weeks before OTA administration with a combined antioxidants, 1.5 mg vitamin E (dl-a-tocopherol)/g diet), sodium selenite (2.0 mg/g diet), ascorbic acid (350 mg/mg diet), CoQ 10 350 mg/g diet, L-carnitine 350 mg/g diet, Zn 291 mg/g diet, Mg 4.5 mg/g diet and N-acetyl cysteine 250 mg or with tamoxifen at a daily dose of diet containing 15 mg TMX (tamoxifen citrate, Sigma Chemical Co, St. Louis, MO, USA)/g food) for periods of one week.
Ochratoxin A administration. OTA (Sigma Chemical Co, St. Louis, MO, USA) was dissolved in seed oil and administered orally at a dose level of 1.8 mg/kg body weight by gastric intubations twice a week for one or two weeks period. Our aims was mainly to produce acute toxicity so the results would be affective. Control animals were given seed oil only. The liver was removed from the first group of animals after one week of OTA administration and after two weeks OTA administration. The hepatic tissue samples were excised and rinsed with ice-cold homogenising buffer (50 mM Tris, 0.1 mM EDTA, pH 7.6). The rest of the tissue samples were frozen in liquid N2 and stored at -85°C.
Histological Examination. On the day the animals were killed, each mouse was weighed, and the liver was dissected, embedded in Cryo-M-bed (Bright Instrument Co), and immediately snap-frozen in liquid nitrogen. Simultaneously, small portions of the fresh liver were immediately fixed in Bouin's solution and embedded in paraffin; sections 5 mm thick were stained with hematoxylin and eosin.
Incidence of Apoptotic Bodies. Hematoxylin-eosin-stained sections were examined for the scoring of apoptotic bodies (41). The number of cells undergoing apoptosis was randomly counted in 100 to 200 high-power microscope fields with a laborlux D microscope (Leitz, Germany). Approximately 10,000-20,000 hepatocytes per mice were counted. Seven mice per group were used. The number of apoptotic bodies was expressed as number per 100 nuclei. Only apoptotic bodies containing nuclear fragments were recorded. Using a unique magnification microscope we were able to observe and differentiate between apoptotic bodies and cell-depressed.
Liver glutathione (GSH) measurement. Immediately after removal, liver tissue samples (0.5 g)from animals treated with OTA for two weeks were homogenized in 10 ml ice-cold homogenising buffer combined with sulphosalicylic acid with two 10 sec. burst of tissue disintegrator at 12,000 r.p.m. The tissue homogenate was used for measuring hepatic reduced glutathione (GSH) content using Ellmans' reagent (42). Briefly, an aliquot (0.2 ml) of hepatic homogenate was combined with 10% sulphosalicylic acid. After centrifugation at 12,000 rpm for two minutes in a Eppendorf centrifuge at 4°C, the supernatant was analysed spectrophotometrically at 412 nm, with 5,5' -dithiobis(2-nitro benzoic acid) in 0.1 M phosphate buffer, pH 8.0, for non-protein thiols.
Statistical analysis. The data were analyzed by analysis of variance, and where statistically significant differences were found, they were then evaluated with a Student's t-test. The comparison made were: control versus deficient, control versus antioxidant-supplemented or tamoxifen, control versus OTA-treated, antioxidant-supplemented or tamoxifen plus OTA versus antioxidant-supplemented or tamoxifen without OTA, OTA-treated versus OTA plus antioxidants or tamoxifen treated (43).
RESULTS
Effect of Ochratoxin A on Liver Tissue. We did a preliminary experiment to study the effect of OTA administration on liver apoptosis at different days after toxin administration in order to identify the most suitable time for the occurrence of apoptotic bodies. Examination of the liver after mice administered a dose of 1.8 mg OTA/kg body weight twice a week for two weeks, disclosed the presence of a difference between hepatocytes of different zones of the acinus, in that an eosinophilic-dense cytoplasm was characteristic of the liver cells located in the periphery of the acinus. Apoptotic bodies were frequently observed in the proximity of the centrilobular vein and in the periphery of the acinus (Figure 1)
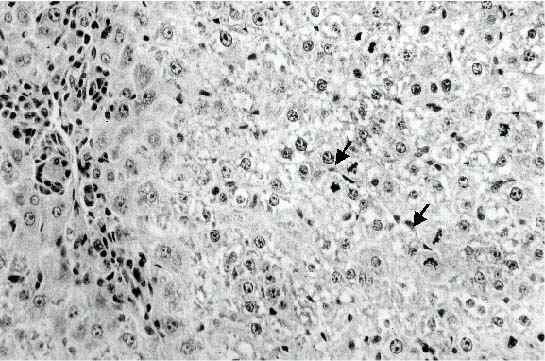
Figure 1: Liver of mouse given 1.8 mg/kg body weight of OTA twice a week for one week. Many apoptotic bodies are present either in clusters in the central area (arrows) or scattered between the surrounding hepatocytes with condensed cytoplasm (Original magnification: x 300)
They were scattered or in small clusters, and as for their localization they were either in the extracellular space or inside the cytoplasm of intact hepatocyte cell. Only little evidence of inflammatory reaction could be observed at one week, no signs of lytic cell necrosis could be detected. At two weeks, an increase in the number of acidophilic globules was observed, they were often found surrounded by mononuclear cells (Figure 2).
A prominent degree of centrilobular necrosis accompanied by inflammatory reaction was also observed in the liver of mice killed at two weeks after OTA administration. Several clusters of acidophilic globules surrounded by inflammatory cells could be observed around the centrilobular vein. Scattered apoptotic bodies were occasionally seen also in the surrounding areas.
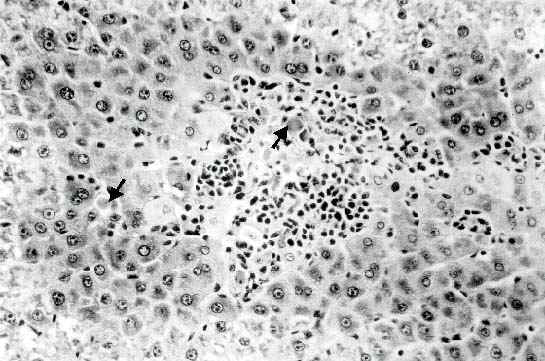
Figure 2: Liver of mouse given 1.8 mg/kg of OTA twice a week for two weeks, centrilobular necrosis with inflammatory reaction can be observed. Centrilobular necrosis with inflammatory reaction is associated to the presence of several mitotic figures (arrows). (Original magnification: x 300).
Although a similar degree of inflammation was observed in the hepatic pericentral areas of mice killed one week after administration of OTA, a reduction in the number of acidophilic globules was apparent at the same time. Several hepatocytes in mitosis were observed in the acinus, suggesting that regeneration was occurring. Thus from these studies it appears that two morphologically distinct types of cell death, namely apoptosis and necrosis, are induced by pretreatment with OTA for two weeks.
Quantitation of Apoptotic Bodies. To quantitate the incidence of apoptotic bodies (44), sections of liver tissue from mice treated with 1.8 mg OTA/kg body weight, for two weeks and then killed were analyzed. As indicated in Figure 3, only a very small number of apoptotic bodies could be detected in control liver (0.009 ± 0.002 ABs/100 nuclei), thus confirming the very low turnover of adult hepatocytes. Although OTA administration did not significantly increase the incidence of apoptotic bodies one week after treatment in liver, a 8.3 fold increase was found at two weeks when compared to control liver.
Antioxidants micronutrients inhibit apoptosis. Effects of the combined antioxidants (CoQ 10, L-carnitine, Zn, Mg, N-acetyl cysteine, vitamin C, vitamin E and selenium) being efficacious, which are known to act as superoxide anion scavengers, were tested on genotoxicity of OTA. Pretreatment of mice by these antioxidants induced a 20% decrease in apoptosis in liver (Figure 3). The results indicate that antioxidants treatment exerted some inhibitory effect on the formation of apoptotic bodies.
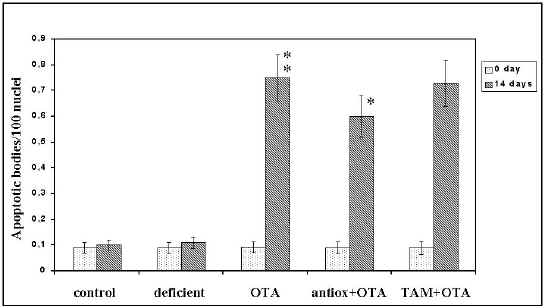
Figure 3: Effect of the antioxidants on the incidence of apoptotic bodies induced by OTA. Mice were given combined antioxidants (CoQ10, L-carnitine, Zn, Mg, vitamin C, vitamin E and selenium) for 10 weeks or tamoxifen for 1 week. Two weeks after OTA administration the animals were killed. (results are mean ± SEM). Significant = P< 0.05; P< 0.01
Effect of the Tamoxifen on OTA-induced Apoptosis. It has been reported that tamoxifen is a membrane antioxidant in that it protects against the potentially carcinogenic free radical intermediates and the products of lipid peroxidation (38). In contrast, it has also been reported that tamoxifen induces apoptosis (45). Therefore, we investigated whether treatment with this chemical could inhibit also OTA-induced apoptosis or whether it would be ineffective. The results obtained indicate that tamoxifen had no significant inhibitory effect on the occurrence of apoptotic bodies induced by OTA (Figure 3).
Glutathione Activity. To determine whether the activity of GSH could decrease during apoptosis that follows administration of OTA for two weeks, the biochemical activity of this enzyme was analyzed. As shown in Figure 4, hepatic glutathione decreased significantly (P<0.01) by 52% at 14 days treatment (19.7±1.8 nmol/mg liver for treated mice compared to 37.7 ±1.9 nmol/mg liver for non treated mice).
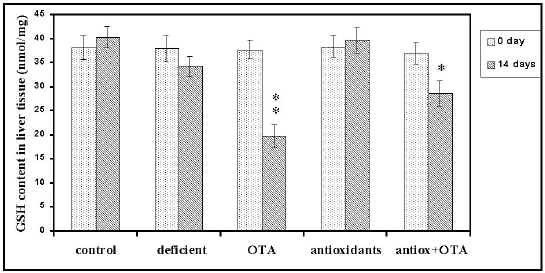
Figure 4: Effects of combined antioxidants pretreatment on OTA- induced liver glutathione depletion (results are mean ± SEM). Significant = P< 0.05; P< 0.01
This finding together with our previous observations indicates that induction of cell death, in the form of apoptosis, may be detected at a biochemical level. The implications of this observation in testing the possible toxicity of the chemical are obvious. Additionally, antioxidants supplementation showed a protective effect (P<0.05) against GSH content depletion (23%) caused by OTA.
DISCUSSION
Cell death takes two distinct forms, necrosis and apoptosis. Although necrosis is considered to be a degenerative phenomenon commonly associated with chemical injury, apoptosis, in contrast, appears to be an active endogenous process implicated in the regulation of normal as well as preneoplastic and neoplastic tissues (46,47,48).
The present study demonstrates that administration of OTA for two weeks induces apoptosis. We believe that the term apoptosis is appropriate at least on a morphologic basis because the type of cell death observed after OTA administration for two weeks was characterized by the finding of all the cardinal morphologic features from the initial chromatin condensation to the intracellular localization of acidophilic globules containing nuclear fragments. Also the absence of any inflammatory response and finally, ultra structural changes specific to apoptosis. In agreement with previous reports (5,11,15,16,17) cell death has been observed after OTA treatment. However, it appears that two distinct types of cell death take place in a sequential fashion. Whether apoptosis may simply represent the initial response of liver cells to a toxic insult that ultimately will lead to necrosis or is a type of cell death that is induced by particular signal is unclear. If the former case is correct, one may postulate that any given toxins may induce both apoptosis and necrosis, depending on the dose and period administered. In the other case, it is possible that not all mycotoxins may be able to induce apoptosis as demonstrated by Ueno et al (17). Additional studies are needed to draw any definitive conclusion on the role of inhibitors of protein synthesis on in vivo. However, OTA resulted in the reduction of protein synthesis in cultured cells (49) induced apoptosis at a significantly low concentration (17). Thus, suppression of the synthesis of protein factor(s) seems to be an important key for the induction of apoptosis. Recently it has been shown that in rats renal tissue, the degeneration increased and only karyomegalic and apoptotic-like cells were observed after OTA administration (50). However, it has been reported that Penicillium polonicum rather than OTA could be implicated in Balkan endemic nephropathy (51).
Another interesting finding of our study is represented by the inefficacy of the tamoxifen to inhibit OTA-induced apoptosis or necrosis. Because tamoxifen, an estrogen antagonist, has also been shown to link to rat liver tumors, endometrial carcinomas (32) and inducing apoptosis (39). It has been postulated that tamoxifen undergoes metabolic bioactivation by cytochrome P-450s to a genotoxic metabolite which is thought to be an epoxide (52). Tamoxifen produces DNA adducts in rat liver (53) and protein adducts in vitro (54). It is hypothesized that tamoxifen can become metabolically activated through selective hydroxylation to form an unstable alkylating species (55). Nevertheless, even though DNA adducts can be formed by human liver microsomes in vitro (56), no practical demonstration of DNA adduct formation has occurred in humans (38). The fact that Tamoxifen did not exert any inhibitory effect on OTA-induced apoptosis raises the question whether the finding that mitogenic stimuli may counteract signals that are responsible for apoptosis may be a general finding or restricted to particular conditions. In particular, it is possible that apoptosis taking place after OTA is the result of a mild injury, and occurring during regression of liver hyperplasia.
A homoeostatic role for apoptosis is often evident also in association with exposure to chemical. In fact, the occurrence of apoptosis in hamster kidney cells and HeLa cells (11,57), human lymphocytes (16), and in HL-60 human promyelotic leukemia cells (17) after exposure to Ochratoxin A has been considered to be part of a regulatory mechanism triggered to restore the original size of the organ after the initial hyperplasia caused by those chemical agents. In all these conditions, apoptosis was the only type of cell death observed, whereas necrosis could not be detected. Co-existence between apoptosis and necrosis was found in the present study after exposure of experimental animals to Ochratoxin A. The finding that apoptosis occurs not only in physiologic circumstances but also under conditions of tissue injury is intriguing and may lead to speculate on common pathways. The mechanisms by which some chemicals may elicit the induction of a form of cell death that appears to be genetically programmed in several physiological conditions is not known.
Glutathione (GSH) one of the most abundant antioxidant in cells has been found to decrease (deplete) during apoptosis. Glutathione has been hypothesized to play a role in the rescue of cells from apoptosis, by buffering an endogenously induced oxidative stress (58). Such changes may result from the oxidation of GSH to GSSG or from the extrusion of intracellular GSH caused by the activation of a distinct GSH- transporter by the apoptotic protease(s) (59). In many cases, both of these mechanisms will probably contribute to GSH depletion during apoptosis. However, it is difficult to establish whether the lack of increase in GSH activity is because OTA-induced apoptosis does not require the activation of a specific gene program, or whether it is a finding unique to some particular property of the toxin used, i.e. it is important to note that OTA is an inhibitor to GSH activity (60). However, it most likely that the hepato-protection afforded by antioxidants pretreatment against OTA hepatotoxicity is mediated by enhancing hepatic GSH status rather than by inhibiting OTA metabolism.
An antioxidant such as GSH also protected in various models of apoptotic cell death, and this phenomenon has been attributed to an interference with apoptotic signalling (61). For example hepatocyte apoptosis as a result of oxidative stress was consistently enhanced by GSH depletion (62). A marked decrease in hepatocyte GSH status has also been reported in cellular necrosis (63). Thus GSH depletion observed in our study raises the possibility of hepatocyte apoptosis and necrosis involvement. However, a definite role for oxidative stress in apoptosis and the role of glutathione in this respect is not clear (64). Our data on the depletion of GSH in livers tissues support the concept that GSH was involved in apoptotic machinery.
Emerging data indicate that reactive oxygen species can serve as potent intracellular second messengers (65). Reactive oxygen species are highly reactive metabolites generated during normal cell metabolism; the cell contains many systems to limit their damaging effects (66), Many agents which induce apoptosis, are either oxidants or stimulators of cellular oxidative metabolisms, whereas many inhibitors of apoptosis show antioxidant activities (35). For example, thiol antioxidants such as N-acetyl cysteine and glutathione completely block the activation-induced cell death of T-cell hybridomas (67). Furthermore, Wedi et al (65) showed significant inhibition of spontaneous eosinophil apoptosis by N-acetyl cysteine and glutathione. In contrast, in neutrophils, N-acetyl cysteine and GSH have been shown to be ineffective in delaying spontaneous apoptosis (68,69).
In this study the antioxidants have shown to have some inhibitors effective on OTA-induced apoptosis. This indicates that to prevent apoptosis effectively, an inhibitor must have several qualities; the ability to neutralize oxygen radicals seems to be an important asset. A number of antioxidants known to inhibit apoptosis are undergoing clinical evaluation. In general, it is recognized that antioxidants play important roles in all aspects of the immune response: phagocyte function, cytokines production, cell-mediated responses, and immunoglobulin production (70). These minerals and vitamins interact and complement each other facilitating immunocompetence at all levels (71). The antioxidant vitamin E and C as well as exogenous catalase have been shown to prevent apoptosis in some system (35). Similarly, transections with selenoenzyme, glutathione peroxidase inhibits apoptotic cell death after growth factor deprivation of an IL-3-dependent cell line (31). A diet supplemented in vitamin E and C seems to reduce the risk of coronary heart disease, a disease in which apoptosis has been demonstrated in the destruction of cardiac cells (72). Both lipophilic free radical scavengers such as vitamin E and hydrophilic scavengers such as ascorbate, glutathione and acetyl cysteine have been shown to have beneficial effects in diabetes (73). Multiple antioxidants, including vitamins C and E, carotenoids, flavonoids, and protein-bound selenium, copper, and zinc may interact additively in biological systems (74).The thiol-based reducing agent N-acetylcysteine (NAC) is a free radical scavenger and might access the endothelial cell thus increasing intracellular glutathione (GSH) stores. Different studies have demonstrated that NAC have the potent antiviral activity (75) and might be a promising compound either for the prevention or the treatment of acute lung damages (76).
In conclusion, taken together, these data could suggest that apoptosis was observed in mice liver after two weeks of OTA administration. The cellular redox state and/or the equilibrium between reactive oxygen species (ROS) generated by OTA and ROS detoxification by the antioxidant could influence the early stage of apoptosis. Thus, ROS are facultative but not obligatory regulators of PCD.
REFERENCES
Hult K, Plestina R, habazin-Novak V, Radic B, Ceovic S. Ochratoxin A in human blood and Balkan endemic nephropathy. Arch Toxicol, 51:313-321,1982.
Purchase IFH. and Theron JJ. The acute toxicity of ochratoxin A to rats. Food Cosmet Toxicol 6:479-483, 1968.
Bendele AM, Carlton WW, Krogh P, Lillehoj EB. Ochratoxin A carcinogenesis in the (C57BL/65 X C3H)F1 mouse. J Natl Cancer Inst 75:733-742, 1985.
Pitt JI. Toxigenic fungi and mycotoxins. British Medical Bulletin. 56:184-192,2000.
Wei X, and Sulik KK. Pathogenesis of craniofacial and body wall malformations induced by ochratoxin A in mice. Am J Med Genet 47:862-871,1993.
Creppy EE, Stormer F, Roschentaler R, Dirheimer G. Effects of two metabolites of ochratoxin A, (4R)-4-hydrochratoxin A and ochratoxin á, on immune response in mice. Infect Immunol 39:1015-1018, 1983.
Bondy GS. and Pestka JJ. Immunomodulation by fungal toxins. Journal of Toxicology & Environmental Health. Part B, Critical Reviews 3:109-143,2000.
Engelbrecht JC, and Purchase IFH. Changes in morphology of cell cultures after treatment with aflatoxin and ochratoxin. S Afr J Sci 43:524-528,1968.
Pfohl-Leszkowicz A, Grosse Y, Kane A, Creppy EE, Dirheimer G. Differential DNA adduct formation and disappearance in three mouse tissues after treatment with the mycotoxin ochratoxin A.. Mutat Res 289:265-273,1993.
Maaroufi K, Pfohl-Leszkowicz A, Achour A, el May M, Grosse Y, Hammami M, Ellouz F, Creppy EE, Bacha H. Ochratoxin A genotoxicity, relation to renal tumors. Archives de 1 Institue Pasteur de Tunis 71(1-2):21-31,1994.
Seegers JC, Lottering M-L, Garlinski PJ. The mycotoxin ochratoxin A causes apoptosis-associated DNA degradation in human lymphocytes. Med Sci Res 22:417-419,1994.
Boorman GA . NTP Technical Report on the Toxicology and Carcinogenesis Studies of Ochratoxin A (CAS No. 303-47-9) in F344/N Rats (Gavage Studies). NIH Publication No. 89-2813,1989, U.S. Department of Health and Human Services, National Institute of Health, Research Triangle park, NC.
Manolova Y, Manolov G, Parvanova L, Petkova-Bocharova T, Castegnaro M, Chernozemsky IN. Induction of characteristic chromosomal aberrations, particularly X-trisomy, in cultured human lymphocytes treated by ochratoxin A, a mycotoxin implicated in Balkan endemic nephropathy. Mutat Res Fund Molecul Mechanis Mutagen 231:143-149,1990.
Bose S, and Sinha SP. Modulation of Ochratoxin-produced genotoxicity in mice by vitamin C. Fd Chem Toxic 32: 533-537, 1994.
Albassam MA, Yong SI, Bhatnagar R, Shrma AK, Prior MG. Histopathologic and electron microscopic studies on the acute toxicity of Ochratoxin A in rats. Vet Pathol 24:427-435,1987.
Seegers JC, Böhmer LH, Kruger MC, Lottering M-L, De Kock. A comparative study of ochratoxin A-induced apoptosis in hamster kidney and HeLa cells. Toxicol Appl Pharmacol 129:1-11,1994.
Ueno Y, Umemori K, Niimi E-C, Tanuma S-I, Nagata S, Sugamata M, Ihara T, Sekijima M, Kawai K-I, Ueno I, Tashiro F. Induction of apoptosis by T-2 toxin and other natural toxins in HL-60 human promyelotic leukemia cells. Natural Toxins 3:129-137,1995.
Vaux DL, Haecker G, Strasser A. An evolutionary perspective on apoptosis. Cell 76:777-781,1994.
Schwartzman RA, and Cidlowski JA.. Apoptosis: the biochemistry and molecular biology of programmed cell death. Endocrine Reviews 14:133-151,1993.
Cohen GM. Sun XM. Snowden RT. Dinsdale D. Skilleter DN. Key morphological features of apoptosis may occur in the absence of internucleosomal DNA fragmentation. Biochemical Journal. 286 :331-334,1992.
Samaha HS. Asher E. Payne CM. Bernstein C. Bernstein H. Evaluation of cell death in EBV-transformed lymphocytes using agarose gel electrophoresis, light microscopy and electron microscopy. I. Induction of classic apoptosis by the bile salt, sodium deoxycholate. Leukemia & Lymphoma 19:95-105,1995.
Furuya S. Mitoma J. Makino A. Hirabayashi Y. Ceramide and its interconvertible metabolite sphingosine function as indispensable lipid factors involved in survival and dendritic differentiation of cerebellar Purkinje cells. J Neurochem 71:366-377,1998.
Schulte-Hermann R. Bursch W. Marian B. Grasl-Kraupp B. Active cell death (apoptosis) and cellular proliferation as indicators of exposure to carcinogens. IARC Scientific Publications (Lyon) 146:273-285,1999.
Pollman MJ, Yamada T, Horiuchi M, Gibbons GH. Vasoactive substances regulate vascular smooth muscle cell apoptosis. Circ Res 79:748-756,1996.
Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science 267:1456-1462,1995.
McConkey DJ and Orrenius S. in Apoptosis: The Molecular Basis of Cell Death (Tome LD and Cope FO, eds),pp.227-246,1991, Cold Spring Harbor Laboratory Press.
Thatte U and Dahanukar S. Apoptosis: clinical relevance and pharmacological manipulation. Drugs 54:511-532,1997.
Meier PJ. Eckhardt U. Schroeder A. Hagenbuch B. Stieger B. Substrate specificity of sinusoidal bile acid and organic anion uptake systems in rat and human liver. Hepatology 26:1667-1677,1997.
Rizzo AF, Atroshi F, Ahotupa M, Sankari S, Elovaara E. Protective effect of antioxidants against free radical-mediated lipid peroxidation induced by DON or T-2 toxin. J Vet Med A 41:81-90,1994.
Hockenbery DM, Oltvai ZN, Yin X-M, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 75:241-251,1993.
Kane DJ, Sarafian TA, Anton R, Hahn H, Cralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science 262:1274-1277,1993.
Coyle JT, and Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262,689-695.
Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B, Kroemer G . Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med 182:367-377, 1995.
Buttke TM, and Sandstrom PA. Oxidative stress as a mediators of apoptosis. Immunol Today 15:7-10,1994.
Wiseman H (1994) Tamoxifen: new membrane mediated mechanisms of action and therapeutic advances. Trends Pharmacol Sci 15,83-88.
Day BW, Tyurin VA, Tyurina YY, Liu M, Facey JA, Carta G, Kisin ER, Dubey RK, Kagan VE. Peroxidase-catalyzed pro- versus antioxidant effects of 4-hydroxytamoxifen: enzyme specificity and biochemical sequelae. Chem Res Toxicol 12:28-37,1999.
Jordan VC. Tamoxifen:Toxicities and drug resistance during the treatment and prevention of breast cancer. Annu Rev Pharmacol Toxicol 35:195-211, 1995.
Atroshi F. Rizzo A. Biese I. Veijalainen P. Saloniemi H. Sankari S. Andersson K. Fumonisin B-1-induced DNA damage in rat liver and spleen: Effects of pretreatment with coenzyme Q(10), L-carnitine, alpha-tocopherol and selenium. Pharm Res 40:459-467, 1999.
Drepper K, Weik H (1972) Versuchstierernährung: Standard und Spezialitäten für warmblütige Versuchstier e. In: Schriftenreihe Versuchstierkunde 1.(P Parey ed),Verlag Parey, Berlin p.9.
Bursch W, Lauer B, Timmermann-Trosiener I, barthel G, Schuppler J, Schulte-Hermann R. Controlled cell death(apoptosis) of normal and putative preneoplastic cells in rat liver following withdrawal of tumor promoters. Carcinogenesis 5:453-458, 1984.
Atroshi F, and Sandholm M. Red blood cell glutathione (GSH) as a marker of milk production in Finnsheep. Res Vet Sci 33:256-259, 1982.
Atroshi F, Rizzo A, Biese I, Lindberg LA, Saloniemi H. Effects of feeding T-2 toxin and deoxynivalenol on DNA and GSH contents of brain of rats supplemented with vitamin E and C and selenium combination. J Anim Physiol a. Anim Nutr 74:157-164,1995.
Kerr JFR, Bishop CJ, Searle J. Apoptosis. In:Recent Advances in Histopathology (PP Anthony, X Macsween, eds),pp 1-15, 1984, Churchill Livingstone, Edinburgh.
Candi E, Melino G, De-Laurenzi V, Piacentini M, Guerrieri P, Spinedi A, Knight RA. Tamoxifen and somatostatin affect tumours by inducing apoptosis. Cancer-Letter 96:141-145,1995.
Wyllie AH, Kerr JFR, Currie AR . Cell death: the significance of apoptosis. Int Rev Cytol 68:251-306, 1980.
Ledda-Columbano GM, Columbano A . Apoptosis and hepatocarcinogenesis. In: Apoptosis: The Molecular Biology of Cell Death (DL Tomei, FO Cope ed), Cold Spring Harbor, NY, Cold Spring Harbor Laboratory Press, 1991.
Kroemer G, Petit P, Zamzami N, Vayssiere JL, Mignotte B. The biochemistry of programmed cell death. FASEB J 9:1277-1287,1995.
Creppy EE, Lorkowsky G, Beck G, Roschenthaler R, Dirheimer G. Combined action of citrinin and ochratoxin A on hepatic tissue culture cells. Toxicol Lett 5:375-380,1980.
Maaroufi K, Zakhama A, Baudrimont I, Achour A, Abid S, Ellouz F, Dhouib S, Creppy EE, Bacha H. Karyomegaly of tubular cells as early stage marker of the nephrotoxicity induced by ochratoxin A in rats. Human & Experimental Toxicol 18: 410-415, 1999.
Mantle PG, Miljkovic A, Udupa V, Dobrota M. Does apoptosis cause renal atrophy in Balkan endemic nephropathy?. Lancet 352: 1118-1119,1998.
Styles JA, Davies A, Lim CK, De Matties F, Stanley LA, White INH, Yuan ZX, Smith LL (1994) Genotoxicity of tamoxifen, tamoxifen epoxide and toremifene in human lymphoblastoid cells containing human cytochrome P450s. Carcinogenesis 15,5-9,1994.
Hard GC, Iatropoulos MJ, Jordan K, Radi L, Kaltenberg OP, Imondi AR, Williams GM. Major differences in the hepatocarcinogenicity and DNA adduct forming ability between toremifene and tamoxifen in female CrL:CD(BR) rats. Cancer Res 53:4534-4541.
Mani C, and Kupfer D. Cytochrome P-450 mediated activation and irreversible binding of the antiestrogen tamoxifen to proteins in rat and human liver:possible involvement of flavin-containing mono-oxygenases in tamoxifen activation. Cancer Res 51:6052-6058, 1991.
Potter GA, McCague R, Jarman M. A mechanistic hypothesis for DNA adduct formation following oxidative metabolism. Carcinogenesis 15:439-442, 1994.
Pathak DN and Bodell WJ. DNA adduct formation by tamoxifen with rat and human liver microsomal activation systems. Carcinogenesis 15:529-532, 1994.
Steyn PS, Vleggaar R, Du Preez NP, Blyth AA, Seegers JC. The in vitro toxicity of analogs of ochratoxin A in monkey kidney epithelial cells. Toxicol Appl Pharmacol 32:198-203, 1975.
Fernandez A, Kiefer J, Fosdick L, McConkey DJ. Oxygen radical production and thiol depletion are required for Ca(2+)--mediated endogenous endonuclease activation in apoptotic thymocytes. J Immunol 155:5133-5139, 1995.
Van den Dobblesteen DJ, Nobel CSI, Schlegel J, Cotgreave IA, Orenius S, Slater AF. Rapid and specific efflux of reduced glutathione during apoptosis induced by anti-Fas/APO-1 antibody.. J Biol Chem 271:15420-15427, 1996.
Chong X, and Rahimtula AD. Alterations in ATP-dependent calcium uptake by rat renal cortex microsomes following ochratoxin A administration in vivo or addition in vitro. Biochem Pharmacol 44:1401-1409, 1995.
Hampton MB, and Orrenius S. Redox regulation of apoptotic cell death. Biofactors 8:1-5, 1998.
Kurose I, Higuchi H, Miura S, Saito H, Watanabe N, Hokari R, Hirokawa M., Takaishi M, Zeki S, Nakamura T, Ebinuma H, Kato S, Ishii H. Oxidative stress-mediated apoptosis of hepatocytes exposed to acute ethanol intoxication. Hepatology 25:368-378, 1997.
63. Gumpricht E, Devereaux MW, Dahl RH, Sokol RJ. Glutathione status of isolated rat hepatocytes affects bile acid-induced cellular necrosis but not apoptosis. Toxic & Appl Pharm 164: 102- 111,2000.
Jacobson MD. Reactive oxygen species and programmed cell death. Trends Biochem Sci 21:83-86, 1996.
Wedi B, Straede J, Wieland B, Kapp A . Eosinophil apoptosis is mediated by stimulators of cellular oxidative metabolisms and inhibited by antioxidants: Involvement of a thiol-sensitive redox regulation in eosinophil cell death. Blood 94:2365-2373, 1999.
Halliwell B, and Gutteridge JMC. Role of free radicals and catalytic metal ions in human disease: An overview. Methods in Enzymology 186:1-85, 1990.
Sandstrom PA, Mannie MD, Buttke TM. Inhibition of activation-induced death in T cell hybridomas by thiol antioxidants: Oxidative stress as a mediator of apoptosis. J Leukoc Biol 55,221-226, 1994.
Watson RW, Rotstein OD, Nathens AB, Dackiw AP, Marshall JC. Thiol-mediated redox regulation of neutrophil apoptosis. Surgery 120: 150-157, 1996.
Watson RW, Rotstein OD, Jimenez M, Parodo J, Marshall JC. Augmented intracellular glutathione inhibits Fas-triggered apoptosis of activated human neutrophils. Blood 89:4175-4181, 1997.
Tengerdy RP. The role of vitamin E in immune response and disease resistance. Annals of the New York Academy of Sciences 587:24-33, 1990.
Chandra RK, and Kumari S. Effects of nutrition on the immune system. Nutrition 10:207-210, 1994.
Riemersma RA,Wood DA,Macintyre CC, Elton RA, Gey KF, Oliver MF. Anti-oxidants and pro-oxidants in coronary heart disease. Lancet 337:677, 1991.
Bloomgarden, ZT. Antioxidants and diabetes. Diabetes Care 20: 670-673,1997.
Packer L. Oxidants, antioxidant nutrients and the athlete. J Sports Sci 15:353-363, 1997.
Kelly GS. Clinical applications of N-acetylcysteine. Alternat Med Rev 3:114-127, 1998.
Domenighetti G. Quattropani C. Schaller MD. Therapeutic use of N-acetyl cysteine in acute lung diseases . Revue des Maladies Respiratoires 16:29-37, 1999
Corresponding author: Faik Atroshi, Department of Clinical Sciences, Pharmacology & Toxicology, Faculty of Veterinary Medicine, P.O. 57, FIN-00014 Helsinki University, Finland. faik.atroshi@helsinki.fi
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps
CSPS Home | JPPS Home | Search | Subscribe to JPPS
|
|
