J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 3(2):268-280, 2000
Regulation of the Multidrug Resistance Genes by Stress Signals
Mahadeo Sukhai and Micheline Piquette-Miller1
Faculty of Pharmacy, University of Toronto, Canada
Abbreviations:
HSF,Heat Shock Factor; IFN,Interferon; IL-1b, Interleukin-1b; IL-6, Interleukin-6; LIF, Leukemia Inhibitory Factor; mdr1,General term for human MDR1 and rodent mdr1a, mdr1b multidrug resistance genes; MDR1, MDR3, Human multidrug resistance genes; mdr1a, mdr1b, mdr2, Murine and rat multidrug resistance genes; MRP, Multidrug resistance associated protein; mrp, Multidrug resistance associated protein genes; PKA,Protein Kinase A; PGP, P-Glycoprotein; TGF, Transforming Growth Factor; TNF,Tumor Necrosis Factor
Manuscript received July 7, 2000, Revised August 14, 2000; Accepted August 31, 2000.
PDF version
Abstract
Transporters in the body play a large role in the distribution and elimination of many clinically important therapeutic substances. Of these, perhaps the one that has been best studied is P-Glycoprotein (PGP), a 170 kDa membrane-bound protein which has been implicated as a primary cause of multidrug-resistance in tumors. An understanding of the physiological regulation of these transporters is key to designing strategies for the improvement of therapeutic efficacy of drugs which are their substrates. To that end, we examine herein the current state of understanding of the molecular regulation of PGP by a variety of endogenous and environmental stimuli which evoke stress responses including cytotoxic agents, heat shock, irradiation, genotoxic stress, inflammation, inflammatory mediators, cytokines and growth factors.Introduction
Transporters in the body play a large role in the distribution and elimination of many clinically important therapeutic agents. P-Glycoprotein (PGP) is a 170 kDa ATP-dependent membrane-bound transporter that is known to confer resistance to a variety of structurally unrelated, clinically important antineoplastic agents (1-3). This phenomenon is generally known as multidrug resistance. PGP is encoded by the mdr1 genes which includes MDR1 in humans and mdr1a and mdr1b in rodents. Overexpression of the mdr1 gene products has been implicated as a primary mechanism of tumor drug resistance (4-6), particularly in tumors arising from tissues which normally express PGP (e.g., the liver, kidney, intestine and blood-brain barrier). Although their physiological function is not clearly defined, the mdr1 gene products are thought to play a role in the protection of organisms against toxic xenobiotics. Studies in mdr1a (-/-) knockout mice have demonstrated increased sensitivity to as well as increased concentrations of xenobiotics such as invermectin, vinblastine, cyclosporin A and digoxin (7,8). Indeed, twenty to fifty fold higher brain concentrations of cyclosporin A and digoxin are found in mdr1a (-/-) knockout mice (9). Gene products of other multidrug resistance gene family members do not play a role in the drug resistant phenotype (10). These include human MDR3 (mdr2 in rodents), which encodes for a phospholipid transporter and SPGP (the sister gene of PGP), which encodes for a hepatic bile salt transporter.
While antineoplastic agents are important substrates of PGP, a variety of other clinically relevant drugs are also transported by PGP. Therefore, in addition to examining PGP overexpression in tumor cells, understanding physiological mechanisms of PGP regulation may help us to explain subject variability in drug disposition. Identification of gene sequences and recent advances in molecular biology have resulted in an explosion of knowledge regarding the genetic regulation of PGP. Thus delineating these regulatory pathways may enable us to predict and manipulate expression of the mdr1 genes in order to improve the clinical effectiveness of PGP substrates.
As PGP functions to protect cells from harmful chemicals and metabolites, it is plausible that these transporters play an important role in the cellular response against stress. Furthermore, numerous "stress-evoking" stimuli have been reported to alter mdr1 gene expression. The promoter regions and upstream regulatory sequences of the human, mouse, hamster and rat mdr1 genes have been identified and characterized (11-14). A summary of what is currently known about the regulatory sequences of the mdr1 genes in human, mouse and rat is presented in Figures 1-3 (compiled from references 15-20). In each of the mdr1 gene sequences, putative binding sites for various "stress" transcription factors are found, including those for AP-1, Sp-1, AP-2, NF-Y, and C/EBPβ (also known as NF-IL6). While the functional significance of these binding sites have yet to be established, it is likely that these sites and transcription factors are the targets of environmental and pathological signaling pathways which evoke the stress response. This review will primarily focus on what is currently known about the effects of heat shock, irradiation, genotoxic stress, inflammation and inflammatory mediators on mdr1 expression and regulation.
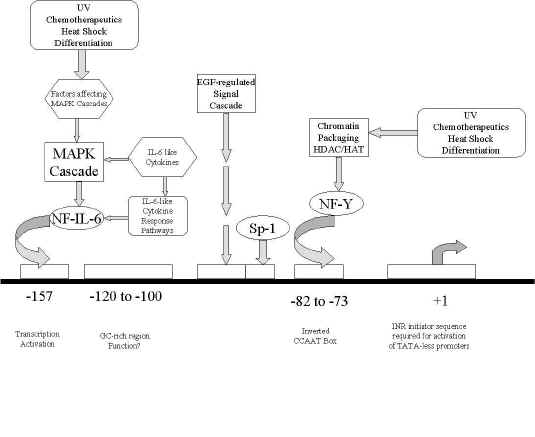
Figure1: schematic of the human MDR1 promoter region, showing the relative locations of transcription factor binding sites, as well as interacting transcription factors and signal transduction pathways.
Mechanisms of mdr1 gene regulation
Although expression of PGP has been frequently examined, regulatory mechanisms of this gene product are complex and still poorly understood. Evidence from numerous studies indicate that expression and activity of PGP can be controlled either pre- or post-transcriptionally by a myriad of environmental influences. For instance, protein kinase C activators which increase PGP activity and drug resistance have been found to enhance mdr1 gene expression via both transcriptional and translational pathways (21). Modulations in protein stability, plasma membrane incorporation, mRNA stability and processing, gene transcription and gene amplification have each been reported for PGP (1-5, 13). Of these, alterations in PGP expression that occur at the level of mRNA are perhaps the most frequently observed (22). Although increased mRNA levels generally occur as a result of enhanced gene transcription rates, prolonged cellular exposure to several cytotoxic drugs have also been reported to induce mdr1 gene overexpression through both gene amplification (4,5) as well as by increased mRNA stability (23). Moreover, mdr1b overexpression in primary cultures of rat hepatocytes occurs primarily due to an increased mRNA stability (24,25). As it is thought that changes in mRNA stability may be tied to cell integrity, it is possible that observed decreases in mdr1b mRNA degradation in cultured cells could result from cellular stress and tissue disruption imposed by collagenase treatments. Furthermore, cytoskeletal disruption of rat hepatocytes by cytochalasin D prevents changes in mdr1b mRNA stability upon culturing (25).
Heat shock
Heat shock proteins are proteins that are synthesized in response to stressful environmental stimuli such as heat. These often include proteins that are thought to help in stabilizing and repairing cell damage. It is likely that efflux transport proteins such as PGP, which are involved in the removal of toxic metabolites and by-products, play an active role in this protective mechanism. Identification of two strong heat shock consensus elements within the human MDR1 gene promoter, as well as an observed in vitro increase in MDR1 mRNA following cellular exposure to high temperature and toxic heavy metals suggest that MDR1 could function as a heat shock gene. It has been shown that basal activity of the MDR1 promoter requires heat shock factor (HSF-) mediated transactivation (26). Indeed, inhibition of the DNA-protein complex formation between HSF and its response element has been found to block MDR1 basal transcription, sensitizing drug resistant cells to anticancer drugs (27). Furthermore, inhibition of protein kinase.
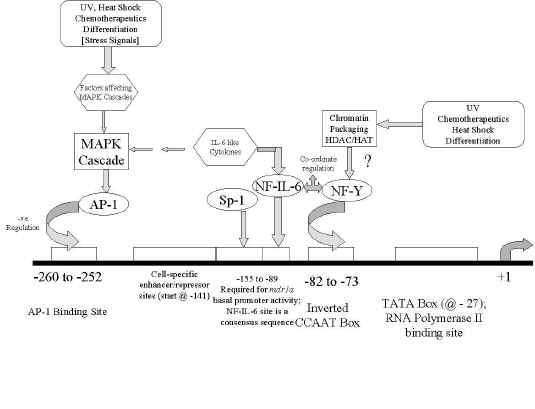
Figure 2: schematic of the murine
mdr1a and mdr1b promoter regions, showing the relative locations of transcription factor binding sites, as well as interacting transcription factors and signal transduction pathways. The information presented here is an amalgam of what is known for both
mdr1a and mdr1b.
A (PKA) suppresses HSF DNA-binding activity as well as reducing expression of the heat shock proteins hsp90 and hsp70 (26). Cells treated with antisense oligonucleotides to both hsp90 and MDR1 have been demonstrated to display vastly decreased PGP half-lives and increased doxorubicin sensitivity to that observed in controls or to that of cells treated with the antisense oligonucleotide to MDR1 alone (28). In these studies hsp90, which could be both co-precipitated as well as co-induced with PGP, was implicated as a possible "chaperone protein" for PGP and is thought to somehow aid in the maintenance of PGP functional activity and protein half-life. Thus suppression of hsp90 expression would likely result in decreased PGP half-life and activity. On the other hand, further experiments conducted by Kim et al (29) have shown that the heat shock element may be involved in alterations of MDR1 transcription rates through pathways that are dependent upon PKA and the raf oncogene. That is, raf activation by heat shock or sodium arsenite, which stimulates the heat shock response, resulted in an induction of PGP activity whereas inhibition of PKA activity using 8-Cl-cAMP, blocked the heat shock potentiation of PGP activity (29). Subsequent studies have established that protein kinase inhibition with 8-Cl-cAMP results in a reduction in MDR1 gene transcription rates (30). Taken together, these data indicate multiple pathways of control of MDR1 expression by cellular pathways that define the heat shock response.
Cells can be made resistant to heat shock (a phenomenon known as "thermotolerance") in the same manner as they can be made drug resistant: Constant and increasing exposure to thermal stresses followed by drug selection conditions. Indeed, in rat hepatoma cells treated in such a manner, there is a correlation between the induction of HSF and the induction of mdr1 gene expression and activity (31). However, in vivo studies contradict these findings, as Vollrath et al (32) presented evidence showing that the rat mdr1 genes are not induced during heat shock. Nevertheless, as a substantial body of evidence exists detailing modulation of mdr1 expression by heat shock, the discrepancies in these studies may be due to difficulties incurred in controlling a heat shock response in a whole animal. Differences in regulation between normal and hepatoma cells, and between species (human and rat) may also play a role. However to date, it seems clear that MDR1 participates as a heat shock response gene in humans. More information in this area may evolve as it is now thought that the heat-shock proteins may play an undefined role in cancer and in the development of drug resistance.
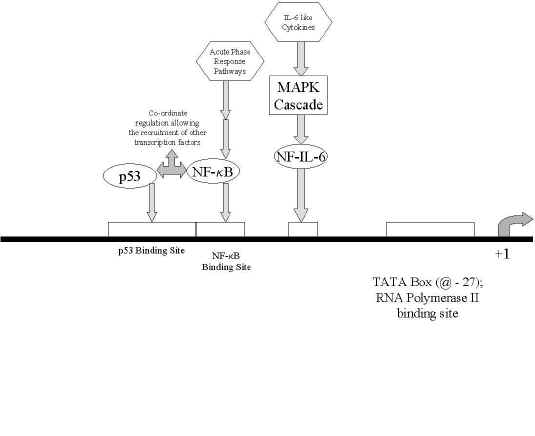
Figure 3: A chematic of the rat mdr1b promoter region, showing the relative locations of transcription factor binding sites, as well as interacting transcription factors and signal transduction pathways. The majority of rat mdr1a promoter regions have yet to be sequenced.
Irradiation
In addition to initiating genetic mutations, ionizing radiation may initiate cellular responses that can ultimately affect mdr1 gene expression. Generally, irradiation has been observed to evoke increased rather than decreased mdr1 expression. It has been demonstrated that induction of MDR1 expression incurred by ultraviolet irradiation results from increased MDR1 gene transcription rates (33,34). Signaling of this induction is thought to primarily occur through a CCAAT box/NF-Y-binding site that is found in the MDR1 promoter (shown in Figure 1). Fractionated X-irradiation has also been found to increase PGP expression in CHO cells due to an increased protein stability and half-life with corresponding decreases in turnover rates (35-38). Indeed, increased PGP half-lives of more than 40 hours are found in irradiated cells as opposed to 17 hours in the control cell populations (36,37). Furthermore, the enhanced stability of PGP reportedly occurs without concomitant increases in mRNA levels (35,38).
Evidence also suggests that the superstructure of chromatin plays a role in transcriptional regulation during UV irradiation. Specifically, the histone acetyltransferases and deacetylases that modulate DNA packaging into histones are believed to be involved. Jin and Scotto (39) reported that incubation of a human carcinoma cell line (SW620) with an inhibitor of histone deacetylase induces a 20-fold increase in MDR1 mRNA levels (39). The authors report that this induction likely occurs through an increased transcription, requiring the sequence from -82 to -73, which contains an inverted CCAAT box element, as point mutations of this sequence were found to abolish promoter response to the histone deacetylase inhibitor. The authors also postulated that ratios of acetyltransferase to deacetylase activities could be important in MDR1 regulation, with hyperacetylation leading to gene activation. Similarly, gel mobility shift assays establishing binding of NF-Y to the inverted CCAAT box and the involvement of NF-Y in intrinsic histone acetyltransferase activity also appear to indicate regulatory mechanisms of human MDR1 gene expression via chromatin acetylation/deacetylation pathways (39). It is unclear whether MDR1 overexpression during radiation exposure is a part of the cellular response to being irradiated or a side effect due to the stimulus. As discussed previously, sequences containing inverted CCAAT boxes were implicated by Ohga et al. (33,34) and by Jin & Scotto (39) in MDR1 induction imposed by UV irradiation. Of note is that this promoter sequence has also been implicated in induction imposed by various stimuli including differentiation (40), heat shock (41) and cytotoxic drugs (33) as this sequence is thought to play a role in maintaining basal MDR1 promoter activity (41,42). This implies that MDR1 may be induced by radiation through a general non-specific cellular response to environmental stress. Nevertheless, as radiation therapy is frequently used concurrent with antineoplastic drugs in cancer treatment, the effect of irradiation on cellular gene expression is certain to receive more attention in the years to come.
Genotoxic stress
Recently, much progress has been made in defining the signal transduction pathways that mediate cellular responses to DNA damage. Contributing to this response are multiple pathways involving alterations of phosphorylation of proteins and transcription factors which occur through several distinct protein kinases (ERK, JNK/SAPK, and p38/HOG1) as well as the tumor suppressor protein p53. In particular, the cyclic AMP responsive transcription factors such as NF- 6B, AP-1, Sp1 and CREB transduce signals in response to protein kinase C activation. To date, several lines of evidence demonstrating a correlation between protein kinase activity and mdr1 expression suggest that activation of cyclic AMP-dependent protein kinases may be involved in induction of the multidrug resistant phenotype in tumor cells (43).
The c-Jun NH2-terminal protein kinase (JNK), a member of a well-characterized mitogen activating protein kinase cascade (44-47) is activated in response to many stressful stimuli including growth factors, phorbol esters, heat shock, UV irradiation, protein synthesis inhibitors, and inflammatory cytokines (46-51). It has been reported that JNK is activated in human carcinoma cells by treatment with a number of different anticancer drugs and this activation of JNK correlates with increased MDR1 expression (44). It is thus believed that JNK may play a role in cellular development of the multidrug resistant phenotype. JNK is known to phosphorylate and activate c-jun, which comprises half of the heterodimeric AP-1 transcription factor (51). It is known now that there are AP-1 binding sites on the promoters of mdr1 genes across species (52) and a positive correlation between AP-1 activation and mdr1 transcription has been reported (53). It is therefore possible that induction of mdr1 expression correlating with JNK activation could be traced to trans-activation by AP-1. Other agents which activate the JNK or protein kinase cascades may also affect mdr1 expression in this manner.
DNA damage is perhaps the best-studied stress that activates p53, and recent data implicate phosphorylation at N-terminal serine residues as critical in this process. It is interesting to note that many members of the mitogen activating protein kinase cascades, because of their roles in controlling cellular growth and proliferation, are oncogenes (c-H-raf and c-H-ras, for example). It has been shown that transformation of rat liver cells with v-H-ras or v-raf oncogenes causes an induction of mdr1/PGP expression (54). In addition to being under the indirect control of those oncogenes, mdr1b in rat hepatoma cells is thought to function as a p53 response gene. Indeed, it has been demonstrated that presence of the p53 response element and the adjacent NF-κB binding site in the mdr1b promoter are both required for basal promoter activity (55,56).
The inflammatory response and cytokines
Induction of an acute inflammatory response in experimental models of inflammation in rats (57) and mice (58) has been demonstrated to decrease the hepatic expression and activity of PGP at the level of mRNA. Findings indicating a down-regulation of the mdr1a, mdr1b and mdr2 genes were obtained in both species with two different inflammation models. These included the turpentine model, which produces a localized inflammatory reaction and the bacterial lipopolysaccharide model, which produces a systemic endotoxemia. Furthermore, experiments in primary hepatocyte cultures treated with lipopolysaccharide also show a reduction in PGP expression and activity (unpublished data). On the other hand, somewhat different results were found in the livers of endotoxemic rat livers by Vos et al (59). His results indicated up-regulation of mdr1b, down-regulation of the bile salt transporter spgp while levels of mdr1a and mdr2 remained unchanged. Although neither quantitative nor statistical analysis of results were presented (59), it is likely that the degree of endotoxemia in the rats may play a role as this may alter the pattern and extent of cytokine release. Indeed, we found that adjuvant-induced arthritis in rats, which is a classical animal model of chronic inflammation, did not significantly alter the hepatic expression of PGP (unpublished data). Furthermore, other studies in the laboratory indicated that renal and intestinal expression of the mdr1 genes remain unchanged in LPS or turpentine-injected rats, suggesting that suppression of the mdr genes during acute inflammation is both a complex and liver-specific phenomenon.
It is well known that the majority of effects seen during an acute inflammatory response are associated with the release of a few of the pro-inflammatory cytokines. In particular, it has been demonstrated that interleukin 1b (IL-1b) and IL-6, and to a lesser extent, tumor necrosis factor (TNF-) a, are the principle mediators involved in controlling the hepatic gene expression of numerous glycoproteins, as well as the cytochrome P450 drug metabolizing enzymes during inflammation. Therefore, it is likely that these mediators are also involved in PGP regulation and the control of mdr gene expression during an inflammatory response. Indeed, in vitro treatments of cultured hepatocytes with recombinant IL-1b and IL-6 elicit dose- and time-dependent reductions in PGP expression and activity (60-62). Results demonstrating decreases in mdr1 mRNA in IL-6 but not IL-1 treated cells (60-62), suggests that IL-1b-mediates effects on PGP expression via post-translational mechanisms, whereas IL-6 likely influences PGP expression by either decreasing mdr1 mRNA stability or reduced transcription rates. Experiments are underway to clarify this matter. In vivo experiments in mice have also demonstrated similar cytokine-mediated effects on PGP and mdr1 expression (58). These data also support the hypothesis that IL-6 is primarily responsible for down-regulation of PGP expression and activity during an acute inflammatory response.
Several studies also indicate that TNF-b, which primarily acts through NF-kB, suppresses mdr1b gene expression. These in vitro studies report down-regulation of PGP protein and MDR1 gene expression as well as enhanced chemosensitivity in continuous human intestinal cell lines treated with TNF-∀ (63-65). A binding site for NF-kB exists on the mdr1b promoter (55) which may implicate the potential involvement of this transcription factor in mdr1b down-regulation. On the other hand, others have also observed a TNF-α mediated induction of mdr1b expression in cultured rat hepatocytes that can be suppressed by addition of the anti-inflammatory agent dexamethasone (66,67). The apparent discrepancies in these studies are likely due to species and TNF treatment differences. Furthermore, as different cell types are unique in their ability to produce and release other cytokines, the use of intestinal or hepatic cells in these studies likely influence cellular exposure to other cytokines which could contribute to their dissimilar findings.
In terms of species differences, although we have observed that the inflammatory response mediates a suppression of PGP in rats (57) and mice (58), this phenomenon has yet to be examined in humans. Several reports indicate a diminished MDR1 gene expression and/or potentiation of chemosensitivity in human colon carcinoma cell lines incubated with a number of these cytokines including IFN-g, TNF-a, IL-2 and leukoregulin (63-65,68-70). While information in this area is limited, IFN-g, TNF-α effects are mediated through an inhibition of MDR1 gene transcription (63,64). Studies with IFN-a have also demonstrated an IFN-α mediated downregulation of MDR1 in a human hepatoma cell line (71). Other cell types have not produced consistent results with cytokine treatments (72). In vivo studies have not yet been conducted however, observations of therapeutic synergism have been reported in patients given combinations of cytotoxic drugs with IFN or TNF (73,74).
While the molecular pathways involved in cytokine-mediated regulation of mdr1 gene expression have not been delineated it is likely that the down-regulation of mdr1/PGP in hepatocytes occurs through inhibition of mdr1 gene transcription. It is felt that the cytokines mediate their effects through unique signal transduction pathways involving only a handful of nuclear transcription factors (NF-6B, C/EBP and APRF) (75). Changes in hepatic protein production during an inflammatory response are thought to be primarily mediated through the nuclear factor NF-IL6 (also known as C/EBPβ) which belongs to the C/EBP transcription factor family (75,76). During an inflammatory response or after exposure to IL-1, IL-6 or TNF, dramatically increased levels of NF-IL6 function as both a positive and negative regulator of transcription within the liver (76). Binding sites for NF-IL6 and other C/EBP transcription factors have been identified on the promoter region of the mdr1 genes (15,16,19). Thus transcription control through members of the C/EBP family such as NF-IL6 may provide a possible cellular signaling pathway by which inflammation and inflammatory mediators suppress PGP expression (20).
Preliminary investigations in our laboratory with Leukemia Inhibitory Factor (LIF) also indicate a complex pattern of mdr1 mRNA suppression (77). It is known that IL-6 and LIF activities are mediated through similar pathways: MAPK activation of NF-IL-6 (C/EBPb) and/or induction of the acute phase response factor through the JAK/STAT pathway (78). However, the mechanisms through which both cytokines act on PGP expression have not yet been elucidated. The anti-proliferative cytokine, transforming growth factor (TGF-) b1, which is released during an acute inflammatory reaction also appears to influence mdr1 gene expression in a complex manner. Long-term exposure to TGF-b1 induces drug resistance by induction of MDR1 mRNA expression (79) whereas short-term exposure to TGF-b1 in glioblastoma cells has been reported to decrease MDR1 expression (80). Initial studies in cultured rat hepatocytes in our laboratory also demonstrated an induction of mdr1b mRNA after 24 hours of exposure to TGF-b1 (77). It is known that TGF-b1 operates through a family of transcription factors and associated proteins known as the Smads, which interact with AP-1 (81-85). As AP-1 binding sites exist in the promoters of members of the mdr1 gene family, this may provide a link to TGF-b1 modulated effects on PGP/mdr1 transcription.
In addition, other growth factors are known to induce and modulate PGP/mdr1 expression. In particular, epidermal growth factor and insulin-like growth factor-1 are both known to induce PGP and mdr1b expression in a time-dependent manner in cultured rat hepatocytes (86). Aside from likely effects on gene transcription, resulting in increased mRNA levels, it is possible that epidermal growth factor could also have an effect on the post-translational regulation of PGP, via phospholipase C and increased phosphorylation of PGP (87). In the case of insulin-like growth factor-1, it is thought that it may induce mdr1 gene expression via a c-H-ras dependent MAPK signaling cascade (88).
To date, the effects of cytokines on PGP expression have not been fully delineated. In addition to possessing complex patterns of induction and inhibition, the cytokines have many overlapping and synergistic effects. Thus it is likely that the cytokines also interact with mdr1 gene expression in a elaborate manner resulting in unique effects dependent upon cytokine concentrations, cell type and species. As many disease states are associated with changes in cytokine secretion there is a need to explore PGP regulation in both health and disease. Further investigation is also necessary, for although cytokine-mediated mechanisms of MDR1 down-regulation have promising usefulness in cancer treatments, available information in this area is limited with many discrepancies reported.
The Multidrug Resistance-Associated Protein (MRP)
The overexpression of other drug efflux transporters such as the multidrug resistance-associated proteins (MRP) is beginning to be recognized as playing an important role in the development of drug resistance in tumors. Much in this area needs to be explored. As several members of the MRP transporter family are also induced by cytotoxic drugs (89-92), the potential for environmental and physiological regulation of these genes also exists. Indeed, this field is still in its infancy and promoter sequences of the mrp genes have not yet been reported. Thus little is known about the signaling pathways that regulate MRP. However, several pathways of MRP gene regulation appear to occur through stimulation by environmental factors. For example, fractionated gamma-irradiation of a human T-cell leukemic cell line reportedly causes a six-fold increase in levels of mrp1 mRNA levels (93, 94). It therefore seems likely that mrp1 is somehow involved in the immediate cellular response to radiation. As mdr1 levels are also increased through irradiation (34,94), induction and overexpression of mdr1 and mrp1 following radiation therapy likely contribute to the clinical development of resistance in tumors. Furthermore, numerous mechanistic links between p53 activity and mrp1 and mdr1 expression have also been reported in drug resistant cells. Wild type p53 reportedly suppresses mrp1 transcription (95) and significant increases in mrp1 mRNA levels are seen in p53 inactivated or mutated cell lines (96). This negative regulatory effect of p53 is thought to occur through effects on the transcription activator Sp-1 (95). Likewise, levels of mdr1a mRNA and PGP are also markedly elevated in p53 inactivated rodent hepatoma cells (97) whereas rat mdr1b gene expression and basal mdr1b promoter activity requires and is induced by wild type p53 activity (56). Conversely, a recent study, indicating a lack of effect of heat shock on mrp1 expression in esophageal cancer cells, may suggest that mrp1 does not function as a heat shock gene (98). However, this evidence is limited and further studies in alternate cell lines are necessary to fully establish whether MRP induction contributes to drug resistance elicited by heat shock treatments.
Interestingly, we as well as others (89, 102) have seen that induction of an acute inflammatory reaction reduces the expression and activity of mrp2 at the level of mRNA. This downregulation of mrp2 likely occurs due to pro-inflammatory cytokine release, as our preliminary studies have observed reductions in mrp2 mRNA and protein expression in mice treated with IL-6, IL-1β or TNF-α (unpublished results). Furthermore, the anti-inflammatory agent, dexamethasone has been shown to prevent inflammation-induced changes in mrp2 expression (99). It appears that other MRP family members may also be regulated through cytokine-mediated pathways as mrp1 mRNA expression is reportedly diminished in human hepatoma cell lines upon interferon and TNF-α treatment (100,101). It will be interesting to see how the field develops in a few years, and to compare more fully the regulation patterns of the mdr and mrp gene families.
Summary
A number of environmental stimuli are known to affect the expression of the mdr1 genes. In humans, it has been suggested that MDR1 may function as a heat shock gene and its expression may be modulated by the numerous agents which trigger these signaling pathways. Ionizing radiation has also been shown to up-regulate mdr1 gene expression across species via a number of mechanisms. Evidence demonstrates UV-irradiation modulates human MDR1 gene transcription through chromatin packaging mechanisms; a novel regulatory pathway first seen with the MDR1 gene. Other stress stimuli including a variety of cytokines and growth factors have also been demonstrated to elicit effects on mdr1 gene expression. Although much has been reported on mdr gene expression, the mechanisms involved in its regulation are still relatively unknown. It is likely that a limited number of transcription factors possessing binding sites on the mdr1 promoters are involved in stress-stimulated regulatory pathways. For instance, mdr1 induction by irradiation appears to be part of a general response system via activation of NF-Y. However, many environmental stress signals also trigger the JNK cascades for AP-1 activation. As the mdr1 gene contains response elements for AP-1 on its promoter sequences, it seems likely that gene expression may be regulated by cellular stress through alternate pathways stimulated by NF-Y and AP-1.
Further studies to confirm the involvement of these transcription factors will need to be done before our understanding of the regulation of PGP under conditions of physiological stress is complete. Nevertheless, it seems likely that cellular stress and PGP expression are closely linked as this transporter plays a crucial role in the protection of cells from toxic products released during environmental stress. As PGP expression is an important in the clinical efficacy of drugs in both normal and malignant cells, it is anticipated that knowledge of regulatory mechanisms may aid in identifying and developing novel therapeutic strategies to modulate mdr1 gene expression.
Acknowledgements
Novel results presented in this review were funded by grants obtained from the Medical Research Council of Canada.
References
- Childs, S. and Ling, V., The MDR superfamily of genes and its biological implications. Important Adv Oncol., 1994:21-36, (1994).
- Fardel, O., Lecureur, V. and Guillouzo, A., The PGP multidrug transporter. Gen. Pharmacol, 27: 1283-91, 1996
- Sharom, F.J., The PGP efflux pump: how does it transport drugs? J. Memb. Biol. 160: 161-75, 1997.
- Riordan, J.R., Deuchars, K., Kartner, N., Alon, N., Trent, J. and Ling, V, Amplification of PGP genes in MDR mammalian cell lines. Nature 316, 817-19, 1985
- Shen, D.-W., Fojo, A., Chin, J.E., Roninson, I.B., Richert, N., Pastan, L and Gottesmann, M.M, Human multidrug-resistant cell lines: increased mdr1 expression can precede gene amplification. Science 232, 643-5, 1986.
- Lum, B.L. and Gosland, M.P. MDR expression in normal tissues. Hematol Oncol. Clin North Am., 9: 319-36, 1995.
- Borst, P. and Schinkel, A.H., Mice with disrupted PGP genes. In S. Gupta and T. Tsuruo, (eds), Multidrug Resistance in Cancer Cells, John Wiley & Sons Ltd. New York 1996
- Borst, P. and Schinkel, A.H., Genetic dissection of the function of mammalian PGPs. Trends Genet., 13, 217-22, 1997.
- Schinkel, A.H., Wagenaar, E., Van Deemter, L., Mol, C.A. and Borst, P, Absence of the mdr1a PGP in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin and cyclosporin A. J Clin Invest., 96: 1698, 1995
- Smit, J.J.M., Schninkel, A.H., Elferink, R.P., et al., Homozygous disruption of the murine mdr2 PGP gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 75:451-62, 1993.
- Ueda, K., Pastan, I. and Gottesmann, M.M, Isolation and sequence of the promoter region of the human MDR (PGP) gene. J Biol Chem, 262, 17432-36, 1987.
- van Groenigen, M., Valentijin, L.J. and Baas, F., Identification of a functional initiator sequence in the human MDR1 promoter. Biochim Biophys Acta, 1172, 138-46, 1993.
- Gottesmann, M.M., Hyrcyne, C.A., Schloenlein, P.V., Germann, U.A. and Pastan, I. Genetic analysis of the multidrug transporter, Annu Rev Genet., 29: 607-64, 1995.
- GenBank Database: http://www.ncbi.nlm.nih.gov/Entrez/nucleotide.html.
- Cohen, D., Yu, L., Rzepka, R. and Horwitz, S.B, Identification of two nuclear protein binding sites and their role in the regulation of the murine mdr1a promoter. DNA Cell Biol, 13, 641-649, 1994.
- Combates, N.J., Rzepka, R.W., Nhen, Y.-N.P. and Cohen, D, NF-IL6, a member of the C/EBP family of transcription factors, binds and trans-activates the human MDR1 gene promoter. J Biol Chem, 269: 29715-19, 1994.
- Raymond, M. and Gros, P, Cell-specific activity of cis-acting regulatory elements in the promoter of the mouse MDR gene mdr1, Mol Cell Biol, 10:6036-40, 1990.
- Sundseth, R., MacDonald, G., Ting, J. and King, A.C, DNA elements recognizing NF-Y and Sp-1 regulate the human MDR promoter. Mol Pharmacol, 51: 963-971, 1997.
- Yu, L., Cohen, D., Piekarz, R.L. and Horwitz, S.B, Three distinct nuclear protein binding sites in the promoter of the murine mdr1b gene, J Biol Chem, 268: 7520-26, 1993.
- Yu, L., Wu, Q., Yang, C.P and Horwitz, S.B, Coordination of transcription factors, NF-Y and C/EBP beta, in the regulation of the mdr1b promoter. Cell Growth Differ 6: 1505-12, 1995.
- Chaudhary PM and Roninson IB, Activation of MDR1 (PGP) gene expression in human cells by protein kinase C agonists, Oncol Res., 4:281-90, 1992.
- Germann, U.A, PGP-a mediator of MDR in tumor cells. Eur J Cancer 32A: 927-944, 1996.
- Lee, C.H., Bradley, G. and Ling, V. Increased PGP mRNA stability in rat liver tumors in vivo. J Cell Physiol, 177:1-12, 1998.
- Gant, T.W., Silverman, J.A., Bisgaard, H.C., Burt, R.K., Marino, P.A. and Thorgeirsson, S.S, Regulation of 2-AAF and methycholanthrene-mediated induction of MDR and cytochrome P4501A gene family expression in primary hepatocyte cultures and rat liver. Mol Carcinogen, 4: 499-509, 1991.
- Lee, C.H., Bradley, G. and Ling, V, Overexpression of the class II PGP gene in primary rat hepatocyte culture: evidence for increased mRNA stability. Cell Growth Differ., 6, 347-354, 1995.
- Kim, S.H., Hur, W.Y., Kang, C.D., Lim, Y.S., Kim, D.W. and Chung, B.S, Involvement of heat shock factor in regulating transcriptional activation of MDR1 gene in multidrug-resistant cells. Cancer Let., 115: 9-14, 1997.
- Kim, S.H., Yeo, G.S., Lim, Y.S., Kang, C.D., Kim, C.M. and Chung, B.S, Suppression of MDR via inhibition of heat shock factor by quercetin in MDR cells. Exp Mol Med, 30: 87-92, 1998.
- Bertram, J., Palfner, K., Hiddemann, W. and Kneba, M, Increase of PGP-mediated multidrug resistance by hsp 90 beta. Anticancer Drugs 7: 838-45, 1996.
- Kim, S.H., Lee, S.H., Kwak, N.H., Kang, C.D. and Chung, B.S. Effect of the activated Raf protein kinase on the human multidrug resistance 1 (MDR1) gene promoter. Cancer Lett., 98: 199-205, 1996.
- Scala, S., Budillon, A., Zhan, Z., Cho-Chung, Y.S., Jefferson, J., Tsokos, M. and Bates, S.E, Downregulation of mdr1 expression by 8-Cl-cAMP in multidrug resistant MCF-7 human breast cancer cells. J Clin Invest, 96, 1026-34, 1995.
- Hever-Szabo, A., Pirity, M., Szathmari, M., Venetianer, A., P-glycoprotein is overespressed and functional in severely heat-shocked hepatoma cells. Anticancer Res., 18: 3045-8, 1998.
- Vollrath, V., Wielandt, A.M., Acuna, C., Duarte, I., Andrade, L. and Chianale, J., Effect of colchicine and heat shock on multidrug resistance gene and PGP expression in rat liver. J Hepatol., 21: 754-63, 1994.
- Ohga, T., Koike, K., Ono, M., Makino, Y., Itagaki, Y., Tanimoto, M., Kuwano, M. and Kohno, K, Role of the human Y box-binding protein YB-1 in cellular sensitivity to the DNA-damaging agents cisplatin, mitomycin C, and ultraviolet light. Cancer Res., 56: 4224-4228, 1996.
- Ohga, T., Uchiumi, T., Makino, Y., Koike, K., Wada, M., Kuwano, M., Kohno, K, Direct involvement of the Y-box binding protein YB-1 in genotoxic stress-induced activation of the human multidrug-resistance 1 gene. J Biol Chem., 273: 5997-6000, 1998.
- Hill, B.T., Deuchars, K., Hosking, L.K., Ling, V. and Whelan, R., Overexpression of PGP in mammalian tumor cell lines after fractionated X irradiation in vitro. J Natl Cancer I., 82:607-12, 1990.
- Hill, B.T., Whelan, R.D.H., Hurst, H.C. and McLean, S., Identification of a distinctive PGP-mediated MDR phenotype in human ovarian carcinoma cells after their in vitro exposure to fractionated X-irradiation. Cancer 73:2990-99, 1994.
- McLean, S. and Hill, B.T., Evidence of post-translational regulation of PGP associated with the expression of a distinctive MDR phenotype in Chinese hamster ovary cells. Eur J Cancer, 29A:2243-48, 1993.
- McLean, S., Hosking, L.K. and Hill, B.T., Dominant expression of MDR after in vitro X-irradiation exposure in intraspecific Chinese hamster ovary hybrid cells. J Natl Cancer I., 85:48-53, 1993.
- Jin, S. and Scotto, J.W., Transcriptional regulation of the MDR1 gene by histone acetyltransferase and deacetylase is mediated by NF-Y. Mol Cell Biol., 18:4377-84, 1998.
- Morrow, C.S., Nakagawa, M., Goldsmith, M.E., Madden, M.J. and Cowan, K.H., Reversible transcriptional activation of mdr1 by sodium butyrate treatment of human colon cancer cells. J Biol Chem., 269:10739-46, 1994.
- Mickley, L.A., Bates, S.E., Richert, N.D., Currier, S.,Tanaka, S., Foss, F., Rosen, N. and Fojo, A., Modulation of the expression of a multidrug resistance gene (mdr-1/PGP) by differentiating agents. J Biol Chem., 264:18031-40, 1989.
- Miyazaki, M., Kohno, K., Uchiumi, T., Tanimura, H., Matsuo, K., Nasu, M. and Kuwano, M., Activation of human MDR1 gene promoter in response to heat shock stress. Biochem Bioph Res Co., 187: 677-84, 1992.
- Rohlff, C. and Glazer, R.I., Regulation of MDR through the cAMP and EGF signaling pathways. Cell Signal., 7: 431-43, 1995.
- Osborn, M.T. and Chambers, T.C. (1996) Role of the stress-activated/c-Jun NH2-terminal protein kinase pathway in the cellular response to adriamycin and other chemotherapeutic drugs. J Biol Chem., 271:30950-5, 1996.
- Lin, A., Minden, A., Martinetto, H., Claret, F.-X., Lange-Carter, C., Mercurio, F., Johnson, G.L. and Karin, M., Identification of a dual specificity kinase that activates the jun kinases and p38-Mpk2. Science 268: 286-90, 1995.
- Yan, M., Dai, T., Deak, J.C., Kyriakis, J.M., Zon, L.I., Woodgett, J.R. and Templeton, D.J., Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature 372: 798-800, 1994.
- Davis, R.J. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem., 268: 14553-6, 1993.
- Cobb, M.H. and Goldsmith, E.J., How MAP kinases are regulated. J Biol Chem., 270: 14843-6, 1995.
- Kyriakis, J.M., Banerjee, P., Nikolakaki, E., Dai, T., Rubie, E.A., Ahmad, M.F., Avruch, J. and Woodgett, J.R., The stress-activated protein kinase subfamily of c-Jun kinases. Nature 369: 156-60, 1994.
- Raingeaud, J., Gupta, S., Rogers, J.S., Dickens, M., Han, J., Ulevitch, R.J. and Davis, R.J., Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem., 270: 7420-6, 1995.
- Hibi, M., Lin, A., Smeal, T., Minden, A. and Karin, M., Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Gene Dev., 7: 2135-48, 1993.
- Ikeguchi, M., Teeter, L.D., Eckersberg, T., Ganapathi, R. and Kuo, M.T., Structural and functional analyses of the promoter of the murine mdr gene mdr3/mdr1a reveal a negative element containing the AP-1 binding site. DNA Cell Biol., 10: 639-49, 1991.
- Volm, M., PGP associated expression of c-fos and c-jun products in human lung carcinomas. Anticancer Res., 13: 375-78, 1993.
- Burt R., Garfield S., Johnson K. and Thorgeirsson S. Transformation of rat liver epithelial cells with v-H-ras or v-far causes expression of MDR1, glutathione-S-transferase-P and increased resistance to cytotoxic chemicals. Carcinogenesis. 12:2329-32, (1988).
- Zhou, G., Song. R. and Kou, M.T., A novel cis-acting element is involved in the promoter activity of the rat mdr1b gene. Cell Growth Differ., 7: 1369-81, 1996.
- Zhou, G. and Kuo, M.T., Wild-type p53-mediated induction of rat mdr1b expression by the anticancer drug daunorubicin. J Biol Chem., 273: 15387-94, 1998.
- Piquette-Miller, M., Pak, A., Kim, H., Anarai, R. and Shahzamiani, A. (1998) Decreased expression and activity of PGP in rat liver during acute inflammation. Pharm Res., 15 706-11,1998.
- Hartmann, G., Kim, H. and Piquette-Miller, M., Suppressive effects of inflammation and pro-inflammatory cytokines on hepatic expression of the mdr genes in mice. Immunopharmacol. "In press", 2000.
- Vos, T.A., Hooiveld, G.J., Koning, H., Childs, S., Meijer, D.K., Moshage, H., Jansen, P.L. and Müller, M. (1998) Up-regulation of the multidrug resistance genes, mrp1 and mdr1b, and down-regulation of the organic anion transporter, mrp2 and the bile salt transporter, spgp, in endotoxemic rat liver. Hepatology, 28: 1637-44
- Sukhai, M., Pak, A., Yong, A. and Piquette-Miller, M., IL-6 decreases PGP expression in cultured rat hepatocytes. AAPS Pharm Sci., 1: S-249, 1999.
- Sukhai, M., Yong, A., and Piquette-Miller, M., Concentration-dependent effects of IL-1 and IL-6 on expression of PGP in cultured hepatocytes. Clin Pharm Ther., 67: 126, 2000.
- Sukhai, M., Yong, A., and Piquette-Miller, M., Decreased expression of PGP in cultured hepatocytes co-incubated with IL-1 and IL-6 Clin Pharm Ther 67: 127, 2000.
- Walther, W. and Stein, U. Influence of cytokines on mdr1 expression in human colon carcinoma cell lines: increased cytotoxicity of MDR relevant drugs. J Cancer Res Clin Oncol., 120: 471-78, 1994.
- Stein, U., Walther, W. and Shoemaker, R.H., Reversal of MDR by transduction of cytokine genes into human colon carcinoma cells. J Natl Cancer I., 88: 1383-92, 1996.
- Stein, U., Waltherm W. and Shoemaker, R.H., Modulation of mdr1 expression by cytokines in human colon carcinoma cells: an approach for reversal of MDR. Brit J Cancer 74: 1384-91, 1996.
- Hirsch-Ernst, K.I., Ziemann, C., Foth, H., Kozian, D., Schmitz-Salue, C. and Kahl, G.F., Induction of mdr1b mRNA and PGP expression by TNF-∀ in primary rat hepatocyte cultures. J Cell Physiol., 176: 506-15, 1998.
- Kreuser E., Wadler S. and Thiel E., Biochemical modulation of cytotoxic drugs by cytokines, Recent Results Cancer Res., 139:371-82, (1995).
- Evans, C.H. and Baker, P.D., Decreased PGP expression in multidrug-sensitive and -resistant human myeloma cells induced by the cytokine leukoregulin. Cancer Res., 52: 5893-99,1992.
- Kang, Y. and Perry, R.R., Effect of IFN-∀ on PGP expression and function and on verapamil modulation of doxorubicin resistance. Cancer Res., 54: 2992-98,1994.
- Walther, W., Stein, U. and Pfeil, D., Gene transfer of human TNF-∀ into glioblastoma cells permits modulation of mdr1 expression and potentiation of chemosensitivity. Int J Cancer 61: 832-39, 1995.
- Takeuchi, A., Kaneko, S., Matsushita, E., Urabe, T., Shimoda, A. and Kobayashi, K., IFN-∀ modulated resistance to cisplatin in three human hepatoma cell lines. J Gastroenterol., 34: 351-8, 1999.
- Bailly, J.D., Pourquier, P., Jaffrezou, J.P., Duchayne, E., Cassar, G., Bordier, C. and Laurent, G., Effect of 5637-conditioned medium and recombinant cytokines on PGP expression in a human GM-CSF-dependent leukemic myeloid cell line. Leukemia 9: 1718-25, 1995.
- Chapekar M.S., Huggett A.C. and Cheng C.; Dexamethasone prevents the growth inhibitory effects of TNF in a rat hepatomas cell line. Biochem. Biophys. Res. Com., 181:1524-31, (1991).
- Wadler S and Schwartz E.L.; Principles in the biomodulation of cytotoxic drugs by interferons., Sem. Oncol. 19:45-8 (1992).
- Vilcek, J. and Le J.; Immunology of cytokines: an introduction. In The Cytokine Handbook 2nd ed. (A. Thomson, ed.) Academic Press, London. Ont. (1994) p1-19.
- Akira S. and Kishimoto T., IL-6 and NF-IL6 in acute phase response and viral infection. Immunol. Rev., 127:25-50, 1992.
- Sukhai M., Yong A. and Piquette-Miller M., Effect of TGF-∃ and LIF on PGP expression in cultured hepatocytes, Submitted for presentation at the 2000 AAPS Annual Meeting, Oct. 29- Nov 2, 2000, Indianapolis, IN.
- Akira, S., IL-6-regulated transcription factors. Int J Biochem Cell Biol., 29: 1401-18, 1997.
- Zhang, Z., Wang, T., Batist, G. and Tsao, M.S., Transforming growth factor beta 1 promotes spontaneous transformation of cultured rat liver epithelial cells. Cancer Res 54: 6122-8, 1994.
- Schluesener, H.J., Multidrug transport in human glioblastoma cells is inhibited by TGF- factors type beta 1, beta 2 and beta 1.2. J Neurosci Res., 28:310-4, 1991.
- Attisano, A. and Wrana, J.L., Mads and Smads in TGF beta signalling. Curr Opin Cell Biol., 10: 188-94, 1998.
- Wrana, J.L., TGF-beta receptors and signaling mechanisms. Mineral Electrolyte Metab., 24: 120-30, 1998.
- Wrana. J.L., TGF-∃ signaling and cirrhosis. Hepatology 29: 1909-10, 1999.
- Wrana, J.L., Regulation of Smad activity. Cell 100: 189-92, 2000.
- Wisdom, R., AP-1: One switch for many signals. Exp Cell Res., 253: 180-5, 1999.
- Hirsch-Ernst, K.I., Ziemann, C., Schmitz-Salue, C., Foth, H. and Kahl, G.F., Modulation of PGP and mdr1b mRNA expression by growth factors in primary rat hepatocyte culture. Biochem Biophys Res Com 215: 179-85, 1995.
- Yang, J.M., Sullivan, G.F. and Hait, W.N., Regulation of the function of PGP by epidermal growth factor through phospholipase C., Biochem Pharmacol., 53: 1597-1604, 1997.
- Guo, Y.S., Jin, G.F., Houston, C.W., Thompson, J.C. and Townsend, C.M., Insulin-like growth factor 1 promotes MDR in MCLM colon carcinoma cells. J Cell Physiol., 175: 141-8, 1998.
- Wielandt, A.M., Vollrath, V., Manzano, M., Miranda, S., Accatino, L. and Chianale, J. Induction of the multispecific organic anion transporter (cMOAT/mrp2) gene and biliary glutathione secretion by the herbicide 2,4,5-trichlorophenoxyacetic acid in the mouse liver. Biochem J, 341:105-11,1999.
- Kaufmann, H.M., Keppler, D., Kartenbeck, J. and Schrenk, D. Induction of cMRP/cMOAT gene expression by cisplatin, 2-acetylaminofluorine or cycloheximide in rat hepatocytes. Hepatology, 26:980-5,1997.
- Kaufmann, H.M., Keppler, D., Gant, T.W. and Schrenk, D. Induction of hepatic mrp2 (cmrp/cmoat) gene expression in nonhuman primates treated with rifampicin or tamoxifen. Arch Toxicol, 72:763-8,1998.
- Oguri, T., Isobe, T., Suzuki, T., Nishio, K., Fujiwara, Y., Katoh, O. and Yamakido, M. Increased expression of the MRP5 gene is associated with exposure to platinum drugs in lung cancer. Int J Cancer, 68:95-100,2000.
- Oosthuizen, M.M., Nel, M.J. and Greyling, D. Heat shock treated oesophageal cancer cells become thermosensitized against anticancer drugs. Anticancer Res, 20:2697-703,2000.
- Harvie, R.M., Davey, M.W. and Davey, R.A. Increased MRP expression is associated with resistance to radiation, anthracyclins and etoposide in cells treated with fractionated gamma-irradiation. Int J Cancer, 73:164-7,1997.
- Wang, Q. and Beck, W.T. Transcriptional suppression of multidrug resistance-associated protein (MRP) gene expression by wild-type p53. Cancer Res, 58:5762-9,1998.
- Sullivan, G.F., Yang, J.L., Vasil, A., Yang, J., Bash-Babula, J. and Hait, W.N. Regulation of expression of the multidrug resistance protein MRP1 by p53 in human prostate cancer cells. J Clin Invest, 105:1261-7,2000.
- Thottassery J.V., Zambetti G.P., Arimori K, Schuetz E.G. and Schuetz J. p53 dependent regulation of MDR1 gene expression causes selective resistance to chemotherapeutic agents. Proc Natl Acad Sci USA, 94:11037-42, 1997.
- Stein, U., Walther, W., Laurencot, C.M., Sceffer, G.L., Scheper, R.J. and Shoemaker, R.H., TNF-∀ and expression of the MDR-associated genes LRP and MRP. J Natl Cancer I., 89: 807-13, 1997.
- Kubitz, R., Wettslein, M., Warskulat, U. and Haussinger, D. Regulation of the multidrug resistance protein 2 in rat liver by lipopolysaccharide and dexamethasone. Gastroenterology, 116:401-10,1999.
- Stein, U., Walther, W., Laurencot, C.M., Sceffer, G.L., Scheper, R.J. and Shoemaker, R.H., TNF-∀ and expression of the MDR-associated genes LRP and MRP. J Natl Cancer I., 89: 807-13, 1997.
- Takeuchi A, Kaneko S, Matsushita E, Urabe T, Shimoda A and Kobayashi K IFN-alpha modulates resistance to cisplatin in three human hepatoma cell lines. J Gastroenterol , 34:351-8, (1999)
- Piquette-Miller, M., ChengYi and Tang W. Endotoxin decreases the over-expression of PGP and MRP2 in AAF-treated rats. Clin Pharm Ther., 67: 159, 2000
Corresponding Author: Dr. Micheline Piquette-Miller, Faculty of Pharmacy, University of Toronto, 19 Russell Street, Toronto, Ontario, Canada, M5S 2S2. m.piquette.miller@utoronto.ca.
JPPS Contents
Published by the Canadian Society for Pharmaceutical Sciences.
Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences.
http://www.ualberta.ca/~csps
CSPS Home | JPPS Home | Search | Subscribe to JPPS |