J Pharm Pharmaceut Sci (www.ualberta.ca/~csps) 2 (2):39-46, 1999
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
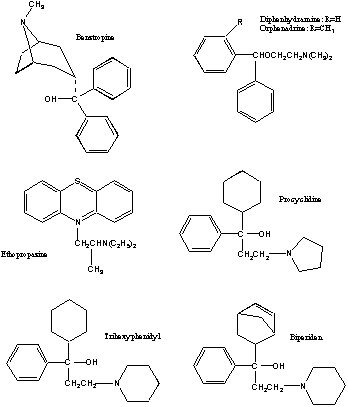
Anticholinergic Drugs Used In Parkinson's Disease: An Overlooked Class Of Drugs From A Pharmacokinetic Perspective. Received manuscript August 9th, 1999; Revised August 20th, 1999; Accepted August 22nd, 1999 Dion R. Brocks1, PDF Version for printing Abstract Anticholinergic drugs were the first pharmacological agents used in the treatment of Parkinson's disease. Although levodopa and other centrally acting dopaminergic agonists have largely supplanted their use, they still have a place in treatment of the disease. As a therapeutic class, there is little pharmacokinetic information available for these drugs, which is inclusive of benztropine, biperiden, diphenhydramine, ethopropazine, orphenadrine, procyclidine and trihexyphenidyl. Pharmacokinetic information is largely restricted to studies involving young health volunteers given single doses. In general, this class of drugs is rapidly absorbed after oral administration to humans. Oral bioavailability is variable between the different drugs, ranging from 30% to over 70%. Each of the drugs appears to possess a large Vd in humans and animals, and distribution to tissues is rapid. The drugs are all characterized by relatively low clearance relative to hepatic blood flow, and appear to be extensively metabolized, primarily to N-dealkylated and hydroxylated metabolites. The available information suggests that excretion of parent drug and metabolite is via the urine and bile. Although the existence of a plasma concentration vs. therapeutic effect relationship has not been explored, there is some evidence suggesting a relationship between concentration and peripheral side effects. Elderly tolerate the drugs less well than do younger patients. There is a notable lack of pharmacokinetic information for these drugs in the elderly. The lack of pharmacokinetic information for multiple dose administration and in the elderly may be a possible hindrance in the safe and effective use of these drugs in patients with Parkinson's disease. Introduction Parkinsons disease (paralysis agitans) is a neurological disorder which principally strikes individuals of middle age and older. In the general population, Parkinson's disease affects approximately 1 in 1000 people (1). It has been estimated that in persons aged 65 years and older, the incidence is approximately 1% (2). From a biochemical perspective, Parkinson's is thought to be caused by degeneration of dopaminergic neurons in the neostriatum, a region of the basal ganglia in the brain important in the regulation of neural impulses which control motor functions (3-5). Although the primary agents used for the pharmacological treatment of Parkinsons disease are those which directly or indirectly act as dopamine agonists, the first drugs used in its treatment were those with anticholinergic activity (4). It was not until the late 1960s that the use of levodopa (L-dopa) for the treatment of Parkinsons disease was established. Although L-dopa has proved to be a monumental advance in the treatment of Parkinsons disease, by itself it is not curative nor sufficient to completely alleviate the symptoms of the disease, which typically include an involuntary resting tremor, muscular weakness, bradykinesia, and muscular rigidity (5). Further, the usefulness of dopamine is limited by a wearing-off phenomenon which appears with prolonged usage (3-5). Although much attention has been directed by researchers and the pharmaceutical industry towards the understanding and development of pharmacological agents which modulate dopaminergic pathways, relatively little effort has been placed towards understanding the pharmacokinetics of those drugs which act at the level of the cholinergic system (1). The anticholinergic drugs used in the treatment of Parkinson's disease (ACP), despite their adjunct role, are nevertheless important agents as they provide an alternate mechanism of action which may alleviate some of the troublesome symptoms of the disease, in particular the involuntary resting tremor. They are used as monotherapy early in the course of the disease, and act synergistically with L-dopa in more advanced stages (3-5). Because ACP are used early in the disease progression, they are potentially helpful in delaying the need for L-dopa treatment. This may allow the patient to derive optimal use of L-dopa later in the course of the disease. In addition, they may allow for a reduction in the dose of L-dopa required in more advanced cases, thus further extending the use of L-dopa (6). The ACP are clinically useful in another respect, as they can diminish the extrapyramidal side effects associated with the use of antipsychotic agents. The purpose of this article is to outline similarities and differences between the ACP, and to highlight deficiencies in the current state of pharmacokinetic knowledge. Mechanism of action of anticholinergics used in Parkinsons disease The ACP are competitive antagonists of muscarinic receptors. Examples used clinically are trihexyphenidyl, benztropine, procyclidine, biperiden, ethopropazine, diphenhydramine and orphenadrine (Figure 1). Each of these drugs are given orally for routine use. Orphenadrine is also available as the citrate salt for the treatment of muscle spasm, and is available as oral and intravenous formulations. The actual mechanism of action for the ACP is not known. It has been theorized that a lesion in the nigra striatum may be responsible for Parkinsons disease, resulting in a reduction in intranigral dopamine concentrations (4). This in turn causes a relative imbalance between the dopaminergic and cholinergic neurological pathways. It has long been believed that anticholinergics can correct this imbalance in less advanced forms of Parkinsons by reducing the degree of neurotransmission mediated by neostriatal acetylcholine (3-5).
Current status of pharmacokinetic knowledge of ACP Currently there is a paucity of information available regarding the pharmacokinetics of the ACP. This has been recognized previously by others. For example, to directly quote from Goodman and Gilmans Pharmacological Basis of Therapeutics 8th edition (7), There is surprisingly little information on the pharmacokinetic properties of these anticholinergic drugs. Although this statement was written a decade ago, it is still germane today. A combination of factors, including onset of newer, more effective antiparkinsons treatments and loss of patent protection, are likely contributors to the lack of pharmacokinetic data for these agents. Another reasonable explanation for a deficiency of pharmacokinetic data is that the doses of several commonly used ACP are very low. This results in correspondingly low plasma concentrations, which may create analytical difficulties in terms of sensitivity (1). Indeed, maximum clinically recommended doses of benztropine mesylate, trihexyphenidyl HCl, and procyclidine HCl are only 6, 10 mg and 20 mg daily, respectively. The structurally related H1-antagonist antihistamine compounds, orphenadrine HCl and diphenhydramine HCl, are generally used in clinically higher doses of at least 100 mg daily. There is also an anticholinergic phenothiazine compound, ethopropazine, which is used at much higher dose levels ranging from 50-600 mg daily. The difference in dose level between the different agents appears to be related to the relative potency of the agents in binding to muscarinic receptors. For example, in muscarinic receptors of rat brain, the IC50 values for atropine, benztropine, trihexyphenidyl, procyclidine, and orphenadrine were found to be 0.0022, 0.018, 0.026, 0.070, and 0.37 mM, respectively (8). Although limited in its quantity and depth, there is some pharmacokinetic data available regarding the anticholinergic agents used in Parkinsons disease. Most of these studies are Phase I evaluations in young healthy volunteers given single doses, or scant reports of pharmacokinetic data from analytical methods papers. There is also some pharmacokinetic information of these agents in animal models. Pharmacokinetic Properties Of The ACP Absorption All of the ACPs (Figure 1) are administered as the HCl salt, with the exception of benztropine; which is given as the mesylate salt. With the exception of benztropine mesylate, the absorption of the other ACPs is relatively quick after oral administration of solid formulations (Table 1). The time to maximal plasma concentrations is generally less than 2.5h. Table 1: Pharmacokinetic indices of anticholinergic antiparkinsons agents in healthy volunteers
In general, the differences in the magnitude of the Cmax attained with these agents after oral administration appear to be related to the dose administered. For example, in humans, Cmax of trihexyphenidyl have been reported to be 10 ng/mL (n=1 subject); 7.15±2.58 ng/ml (n=8 subjects), and approximately 50 ng/mL (n=2 subjects) after single oral doses of 2, 4, and 15 mg, respectively, of trihexyphenidyl HCl (9-11). After a single 2 mg oral dose of benztropine mesylate (12) the observed mean Cmax was only approximately 1.5 ng/mL (n = 5 subjects). Some of these concentrations were measured using radioimmunoassay techniques (10,12), which although sufficiently sensitive, nevertheless leaves open some question regarding assay specificity. Biperiden reaches a Cmax of approximately 4 ng/mL after single oral doses of 4 mg of the HCl salt (13). The plasma concentrations of orphenadrine are much higher, with a mean ± SD of 147±39 ng/mL being attained after single oral doses of 100 mg of the HCl salt (14). Procyclidine (Table 1) provides something of an exception to trihexyphenidyl, biperiden and benztropine, since a single oral dose of 10 mg yields relatively high mean plasma concentrations of 113 ng/mL (15). This Cmax is similar to orphenadrine when given at a higher dose level of 100 mg (14). There is no published information available regarding the pharmacokinetic properties of the phenothiazine derivative, ethopropazine, in humans. The absolute bioavailabilities (F) of all of the ACP have not been reported in humans. Procyclidine, biperiden and diphenhydramine have absolute oral bioavailabilities of 0.75, 0.33 and 0.72, respectively (13,15-16). In the rat ethopropazine has a very low oral F of <5% (17). Intestine has been reported to be the site of absorption of orphenadrine in rats (18). Distribution For each of these drugs in which iv doses have been administered, a Vd exceeding total body water has been reported (Table 1). In the post-distributive phase the plasma concentrations to ACP appear to decline slowly, resulting in relatively long terminal t½ values (Table 1). Relatively low values of plasma CL and Vd (Table 1) appear to be responsible for the relatively high procyclidine plasma concentrations attained relative to other ACP (15). The tissue distribution of some ACP has been observed in laboratory animals. As expected based on the Vd, the tissue:plasma concentrations of these drugs has been reported to be large. In rats, brain:plasma biperiden AUC ratios of 7-12 have been reported (19). Recently it was reported that brain:plasma AUC ratios of 6-8 are present for ethopropazine enantiomers in different regions of brain (20). The heart:plasma concentration ratio was ~3 for ethopropazine in rat (20). The uptake of biperiden, orphenadrine and ethopropazine into the intended site of action, brain, is rapid. After short iv infusion of biperiden to rats, maximal brain concentrations were obtained within 3-10 min (19). After iv bolus doses, ethopropazine tmax in brain tissues is less than 30 min in brain (20). Radiolabelled orphenadrine and its major N-demethylated metabolite, tofenacine, attained maximal concentrations in rat brain in less than 15 min and by 60 min, respectively, after intraperitoneal administration (21). The high concentrations of these drugs in brain tissue may be related to their high lipophilicity. Brain tissues possess high concentrations of lipophilic biomolecules such as sphingomyelin and phosphatidyl choline (22), which might serve to promote the uptake of the ACP. A recent paper has indicated that high tissue uptake of trihexyphenidyl and biperiden were facilitated by intralysosomal uptake of drug (23). The extent of plasma protein binding of ACP is poorly understood in humans. In the rat, ethopropazine is extensively bound (>95%), and nonlinearity was observed in its binding (20) and pharmacokinetics (17). Orphenadrine and biperiden are approximately 90% bound to plasma proteins in animal species (24-26). Metabolism and Excretion In general, all of the ACP appear to be extensively metabolized in humans and animal species. They possess an aliphatic tertiary amine group that is amenable to N-dealkylation and N-oxidation (18, 27-31). Hydroxylation of the alicyclic groups has been reported for the structurally similar compounds procyclidine and trihexyphenidyl (28-30). Benztropine has been reported to undergo N-oxidation, N-dealkylation and ring hydroxylation in rat (31). The CL of the ACP is low in relation to hepatic blood flow (Table 1). Hence, a high hepatic first-pass effect would not be anticipated after oral administration. Several of the drugs in this class are chiral. In humans, stereoselectivity in metabolism has not been formally assessed for any of these compounds. It is known that plasma concentrations of ethopropazine enantiomers are nonstereoselective in the rat (20). The N-demethylated metabolite of orphenadrine, N-demethylorphenadrine, appears to interfere with the metabolism of the parent drug (14). Orphenadrine is known to interfere with P-450 isoenzymes (32). Both urine and bile have been shown to be pathways of elimination of unchanged ACPs and their metabolites (17, 24, 27, 33). Therapeutic Considerations Current guidelines for dosage titration Dosage regimens for ACP are largely empirical in nature, dose titration being dependent on the degree of symptom alleviation and occurrence of dose-limiting side effects, which are primarily a manifestation of peripheral anticholinergic activity (e.g. dry mouth, blurred vision, constipation). Although a few attempts have been made to determine the existence or nature of a plasma concentration vs. toxicological effect relationship after single doses (13, 15, 34), no such data are available for therapeutic effects in Parkinson's disease (e.g. reduction in tremor). Changes in salivary flow and in pupil diameter changes appear to correlate with plasma concentrations of biperiden and procyclidine after both iv and oral administration (13, 15, 34). The relationship between serum trihexyphenidyl concentrations and therapeutic activity has been studied in patients with torsion dyskinesia (35). No relationship was seen between therapeutic effect and serum concentration, although a relationship was present between age and side effects. This study was deficient in several respects. At the time it was believed that the t½ of trihexyphenidyl was 3.5 h (36). Later work using more sensitive assay methodology has indicated that the t½ is a much longer value of 33 h (10). Given that the drug follows multicompartmental kinetics (10), the earlier study (36), which used a less sensitive assay, may have actually reported a distribution phase as being the terminal phase t½.. Furthermore, doses and intervals were not standardized and the number of patients studied was quite small. Consideration of dosage adjustment in the elderly The ACP class of drugs is not as well tolerated in elderly patients as in younger patients. Elderly patients seem to be more susceptible to the peripheral anticholinergic side effects than younger patients (e.g. urinary retention, blurred vision, constipation, tachycardia), and to centrally-mediated effects such as mental confusion (3-5). For this reason, some physicians have recommended that ACP be avoided in elderly patients in favor of L-dopa or other dopaminergic drugs (3). Although this poor tolerability of the elderly towards anticholinergic drugs is a clinically important observation, it is rather surprising that there are only a few reports in the literature dealing with the pharmacokinetics of the ACP in the elderly. This is particularly noticeable in that Parkinsons disease is a condition that primarily afflicts middle aged and elderly patients. It is known that some of these drugs (e.g. orphenadrine) are metabolized to an appreciable extent (27), and that the aging process may be associated with a reduction in liver mass, blood flow, and in the degree of mixed function oxidase enzyme activity (37). The ACP also appear as a class to possess a Vd in excess of total body water (Table 1). After intravenous dosing, the Vd of procyclidine, diphenhydramine and biperiden are 1.1, 4.5 and 24 L/kg, respectively (Table 1). Judging from their values of Vd/F and assuming moderate to complete bioavailability, other ACP (orphenadrine and trihexyphenidyl) also appear to possess a relatively high Vd after oral dosing (10,14,16). Elderly patients have a larger composition of body fat than do younger persons, and thus may have a correspondingly higher volume of distribution compared to younger persons (37). To illustrate this point, in one study involving the disposition of intravenously administered biperiden in the rat, aged rats had an 80% higher volume of distribution at steady-state than young rats, and a CL 39% lower than younger rats (38). The net result was both higher plasma and brain concentrations of biperiden in the aged animals. Assuming that these findings can be extrapolated to other anticholinergics and between species, the lower tolerability of elderly patients for the anticholinergics may be related to pharmacokinetic differences. In humans, age-related differences in the pharmacokinetics of diphenhydramine have been examined. In an elderly group of subjects (Table 1), there was a significantly greater AUC and t½, and lower CL/F compared to a group of young subjects (39). The Cmax was also higher in the elderly subjects compared to the younger subjects. Perhaps surprisingly, the authors found that the Vd of diphenhydramine decreased with advancing age. In contrast to these results, another study reported no noticeable difference in Cmax or AUC between an older group of subjects and a young adult group (40). These two studies differed in that the older cohort studied by Simons et al. (39) were 5 y older than that studied by Scavone and coworkers (40). Further, there was a higher dosage utilized in the study by Simons et al. (39), which perhaps permitted better characterization of the terminal phase t½. Chirality and the antiparkinsons anticholinergic drugs An important consideration pertaining to the pharmacokinetics of the ACPs is that most (trihexyphenidyl, procyclidine, ethopropazine, biperiden, orphenadrine) are chiral and are used clinically as the racemate. With the exception of ethopropazine in the rat (20), there is no published information on the stereospecific pharmacokinetics of these agents. This complicates a possible assessment of their pharmacodynamic and pharmacokinetic properties. It has been demonstrated that for drugs with stereoselective pharmacodynamics, a consideration of the pharmacokinetics of the enantiomers is necessary to accurately define the relationship between plasma concentrations and pharmacological effect (41-43). In fact, for a number of different muscarinic receptor subtypes, the enantiomers of some of the anticholinergics have recently been shown to possess marked stereoselectivity in terms of receptor binding affinities. As examples, the R-enantiomer of trihexyphenidyl possesses up to 427-fold greater affinity than the S-enantiomer for rat striatal muscarinic receptors (44-45). In five cloned human muscarinic receptor subtypes, the R-enantiomer possesses 69 to 525-fold greater binding affinities than its antipode (46). With respect to procyclidine; its R-enantiomer possesses up to 126-fold greater affinity than its antipode for striatal muscarinic receptors (45). Despite these pharmacological differences, the degree of stereoselectivity seen when procyclidine R-enantiomers were administered intraperitoneally or intracerebroventricularly in a mouse model of 6-hydroxydopamine was less than expected based on the binding affinities of the R-enantiomers (47). Stereoselectivity in metabolism of the R-enantiomers was mentioned as a possible cause of this finding. To our knowledge, there are no published reports of the relative pharmacological potency of the R-enantiomers of orphenadrine or ethopropazine. The only published pharmacokinetic information regarding the stereospecific pharmacokinetics of chiral anticholinergic-antiparkinsons agents involves ethopropazine in the rat (20). No stereoselectivity was found in plasma or tissue concentrations, or in the plasma protein binding of the drug. Data from humans is not available. Conclusion The ACP class of drugs has been in clinical use for decades. Even though treatment options have expanded and ACP use is diminished, the drugs are still a part of the pharmacological armamentarium in treatment of the disease. Although newer, more effective treatment options are available for treatment of Parkinson's disease, it is likely that the use of ACPs will persist for the foreseeable future on a worldwide basis. Currently, serious gaps are present in the understanding of the pharmacokinetics of these agents. With the exception of benztropine, assay methodology for ACPs has improved to the point where meaningful pharmacokinetic studies are possible. Little is known of the pharmacokinetics properties of these drugs with repeated dosing to steady-state. The possible relevance of this is best underscored by the findings of nonlinearity of ethopropazine in pharmacokinetics and plasma protein binding in the rat, and by the possibility of a metabolite of orphenadrine which appears to impede metabolism of parent drug. More investigations are definitely required to better understand the cause of the poor tolerability of these drugs in elderly patients. Acknowledgements This work was funded in part by a grant from the Health Services Utilization and Research Commission of Saskatchewan (HSURC). References 1. Contin, M., Riva, R., Albani, F., and Baruzzi, A., Pharmacokinetic optimization in the treatment of Parkinson's disease. Clin Pharmacokinet, 30, 463-481, 1996. 2. Tanner, C.M. Epidemiology of Parkinson's disease. Neurol Clin, 10:317-29, 1992. 3. Sweeney, P.J., Parkinsons disease: Managing symptoms and preserving function. Geriatrics, 1995 (Sep), 50:24-31. 4. Standaert, D.G. and Young, A.B. Treatment of central nervous system degenerative disorders. In Goodman and Gilmans the Pharmacological Basis of Therapeutics, 9th edn,, Hardman, J.G., Limbird, L.E., Molinoff, P.B., Ruddon, R.W. and Goodman Gilman, A. (Eds), McGraw-Hill, New York, 1995, pp. 503-519. 5. Olanow, C.W. and Koller, W.C. An algorithm (decision tree) for the management of Parkinson's disease. Neurology, 50(Suppl. 3):S1-S57, 1998. 6. Bassi, S., Albizzati, M.G., Calloni, E., Sbacchi, M. and Frattola, L. Treatment of Parkinsons disease with orphenadrine alone and in combination with L-dopa. Br J Clin Pract, 40: 273-275, 1986. 7. Cedarbaum, J.M. and Schleifer, L.S., Drugs for Parkinson's disease, spasticity, and acute muscle spasms. In Goodman and Gilmans the Pharmacological Basis of Therapeutics, 8th edn, A. Goodman Gilman, Rall, T.W., Niees, A.S., Taylor, P. (Eds), Permagon, New York, 1990, pp. 463-484. 8. Syvälahti, E.K.G., Kunelius, R. and Lauren, L. Effects of antiparkinsonian drugs on muscarinic receptor binding in rat brain, heart and lung. Pharmacol Toxicol, 62: 90-94, 1988. 9. Owen, J.A., Sribney, M., Lawson, J.S., Delva, N. and Letemendia, F.J.J., Capillary gas chromatography of trihexyphenidyl, procyclidine, and cycrimine in biological fluids. J Chromatogr, 494:135-142, 1989. 10. He, H., McKay, G., Wirshing, B. and Midha, K.K. Development and application of a specific and sensitive radioimmunoassay for trihexyphenidyl to a pharmacokinetic study in humans. J Pharm Sci, 84: 561-567, 1995. 11. Desage, M., Rousseau-tsangaris, M., Lecompte, D. and Brazier JL. Quantitation of trihexyphenidyl from plasma using a mass-selective detector and electron-impact ionization. J Chromatogr, 571: 250-256, 1991. 12. He, H., McKay, G. and Midha, K.K. Development of a sensitive and specific radioimmunoassay for benztropine. J Pharm Sci, 82: 1027-1032, 1993. 13. Grimaldi, R., Perucca, E., Ruberto, G., Gelmi, C., Trimarchi, F., Hollman, M. and Crema, A. Pharmacokinetic and pharmacodynamic studies following the intravenous and oral administration of the antiparkinsonian drug biperiden to normal subjects. Eur J Clin Pharmacol, 29: 735-737, 1986. 14. Labout, J.J.M., Thijssen, C.T., Keijser, G.G.J. and Hespe, W. Difference between single and multiple dose pharmacokinetics of orphenadrine hydrochloride in man. Eur J Clin Pharmacol, 21: 343-350, 1982. 15. Whiteman, P.D., Fowles, A.S.E., Hamilton, M.J., Peck, A.W., Bye, A. and Webster, A. Pharmacokinetics of procyclidine in man. Eur J Clin Pharmacol, 28: 79-83, 1985; 16. Blyden, G.T., Greenblatt, D.J., Scavone, J.M. and Shader, R.I. Pharmacokinetics of diphenhydramine and a demethylated metabolite following intravenous and oral administration. J Clin Pharmacol, 26: 529-533, 1986 17. Maboudian-Esfahani, M. and Brocks, D.R., Pharmacokinetics of ethopropazine in the rat after oral and intravenous administration. Biopharm Drug Dispos, 20: 159-163, 1999. 18. Hespe, W., De Roos, A.M. and Nauta, W.TH., Investigation into the metabolic fate of orphenadrine hydrochloride after oral administration to male rats. Arch Int Pharmacodyn, 156: 180-200, 1965. 19. Yokogawa, K., Nakashima, E., Ichimura J., Hasegawa, M., Kido, H., Ichimura, and F., Brain regional pharmacokinetics of biperiden in rats. Biopharm Drug Dispos, 13: 131-140, 1992. 20. Maboudian-Esfahani, M. and Brocks, D.R., Disposition of ethopropazine enantiomers in the rat: Tissue distribution and plasma protein binding. J Pharm Pharmaceu Sci (www.ualberta.ca/~csps), 2: 23-29, 1999. 21. Roozemond, R.C., Hespe, W. and Nauta, W.Th.. The concentrations of orphenadrine and its N-demethylated derivatives in rat brain, after intraperitoneal administration of orphenadrine and tofenacine. Int J Neuropharmacol, 7: 293-300, 1968. 22. Lehninger, A.L., Nelson, D.L. and Cox, M.M., Biological membranes and transport. In Principles of Biochemistry, 2nd ed., Worth Publishers: New York, 1993, pp 269-297. 23. Ishizaki, J., Yokogawa, K., Nakashima, E., Ohkuma, S. and Ichimura, F., Influence of ammonium chloride on the tissue distribution of anticholinergic drugs in rats. J Pharm Pharmacol, 50:761-766, 1998. 24. Ellison, T. Metabolic studies of 3H-orphenadrine citrate in the rat, dog and rhesus monkey. Arch Int Pharmacodyn, 195: 233-230, 1972. 25. Nakashima, E., Ishizaki, J., Takeda, M., Matsushita, R., Yokogawa, K. and Ichimura, F. Pharmacokinetics of anticholinergic drugs and brain muscarinic receptor alterations in streptozotocin diabetic rats. Biopharm Drug Dispos, 14: 673-684, 1993. 26. Nakashima, E., Yokogawa, Ichimura, F., Hashimoto, T, Yamana, T. and Tsuji, A. Effects of fasting on biperiden pharmacokinetics in the rat. J Pharm Sci, 76: 10-13, 1987. 27. Ellison, T., Snyder, A., Bolger, J. and Okun, R. Metabolism of orphenadrine citrate in man. J Pharmacol Exp Ther, 176: 284-295, 1971. 28. Paeme, G., Van Bossuyt, H. and Vercruysse, A. Phenobarbital induction of procyclidine metabolism: in vitro study. Eur J Drug Metabol Pharmacokinet, 7: 229-231, 1982. 29. Paeme, G., Grimee, R. and Vercruyesse, A. Isolation and identification of eight procyclidine metabolites from rat urine. Eur J Drug Metabol Pharmacokinet, 9: 103-108, 1984. 30. Nation, R.L., Triggs, E.J. and Vine, J. Metabolism and urinary excretion of benzhexol in humans. Xenobiotica, 8: 165-169, 1978. 31. He, H., McKay, G. and Midha, K.K. Phase I and II metabolites of benztropine in rat urine and bile. Xenobiotica, 25: 857-72, 1995. 32. Reidy, G.F., Mehta, I. and Murray, M. Inhibition of oxidative drug metabolism by orphenadrine: In vitro and in vivo evidence for isozyme-specific complexation of cytochrome P-450 and inhibition kinetics. Mol Pharmacol, 35: 736-743, 1989. 33. Hespe, W. and Kafoe, W.F. Aspects of the biliary excretion of orphenadrine and its N-demethylated derivative, tofenacine, in the rat. Eur J Pharmacol, 13: 113-122, 1970. 34. Hollman, M., Brode, E., Greger, G., Muller-Peltzer, H. and Wetzelsberger, N. Biperiden effects and plasma levels in volunteers. Eur J Clin Pharmacol, 27: 619-621, 1984. 35. Burke, R.E. and Fahn, S. Serum trihexyphenidyl levels in the treatment of torsion dystonia. Neurology, 35: 1066-1069, 1985. 36. Burke, R.E. and Fahn, S. Pharmacokinetics of trihexyphenidyl after short-term and long-term administration to dystonic patients, Ann Neurol, 18: 35-40, 1985. 37. Massoud, N. Pharmacokinetic considerations in geriatric patients. In: Benet, L.Z., Massoud, N. and Gambertoglio, J.G., Ed. Pharmacokinetic Basis for Drug Treatment. New York: Raven, 1984, pp 283-310. 38. Yokogawa, K., Nakashima, E. and Ichimura, F. Effect of fat tissue volume on the distribution kinetics of biperiden as a function of age in rats. Drug Metabol Dispos, 18: 258-263, 1990. 39. Simons, K.J., Watson, W.T., Martin, T.J., Chen, X.Y. and Simons, F.E. Diphenhydramine: pharmacokinetics and pharmacodynamics in elderly adults, young adults, and children. J Clin Pharmacol, 30: 665-671, 1990. 40. Scavone, J.M., Greenblatt, D.J., Hermatz, J.S., Engelhardt, N. and Shader, R.I. Pharmacokinetics and pharmacydynamics of diphenhydramine 25 mg in young and elderly volunteers. J Clin Pharmacol, 38: 603-609, 1998. 41. Jamali, F., Mehvar, R. and Pasutto, F.M. Enantioselective aspects of drug action and disposition: therapeutic pitfalls. J Pharm Sci, 78: 695-715, 1989. 42. Mehvar, R. Stereochemical considerations in the pharmacodynamic modelling of chiral drugs. J Pharm Sci, 81:199-200, 1992. 43. Brocks, D.R. and Jamali, F. Stereochemical aspects of pharmacotherapy. Pharmacother, 15: 551-564, 1995. 44. Onali, P., Aasen, A. and Olianas, M.C., Antagonism by (R)- and (S)-trihexyphenidyl of muscarinic stimulation of adenylyl cyclase in rat olfactory bulb and inhibition in striatum and heart. Br J Pharmacol, 113: 775-780, 1994. 45. Waelbroeck, M., Camus, J., Tastenoy, M., Mutschler, E., Strohmann, C., Tacke, R., Schjelderup, L., Aasen, A., Lambrecht, G. and Christophe, J., Stereoselective interaction of procyclidine, hexahydro-difenidol, hexbutinol and oxyphencyclimine and of related antagonists, with four muscarinic receptors. Eur J Pharmacol, 227: 33-42, 1992. 46. Dorje, F., Wess, J., Lambrecht, G., Tacke, R., Mutschler, E. and Brann, M.R., Antagonist binding profiles of five cloned human muscarinic receptor subtypes. J Pharmacol Exp Ther, 256: 727-733, 1991. 47. Barlow, R.B., Dawbarn, D. and Pycock, C.J., A comparison of stereospecificity at central and peripheral "muscarine-sensitive" acetylcholine receptors: observations with the enantiomeric forms of procyclidine and tricyclamol. Br J Pharmacol, 72: 277-280, 1981. Corresponding
author:
Dion R. Brocks, Western University of Health Sciences, College of
Pharmacy, 309 E 2nd Street, College Plaza, Pomona, California, USA,
91766-1854, e-mail: dbrocks@westernu.edu Published by the Canadian Society for Pharmaceutical Sciences. Copyright © 1999 by the Canadian Society for Pharmaceutical Sciences. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|