J Pharm Pharmaceut Sci (www.cspscanada.org) 10(3):319-331, 2007
Lipid Excipients Peceol and Gelucire 44/14 decrease P-glycoprotein mediated efflux of Rhodamine 123 partially due to modifying P-glycoprotein protein expression within Caco-2 Cells.
Kristina Sachs-Barrable, Andrea Thamboo, Stephen D. Lee and Kishor M. Wasan
Division of Pharmaceutics and Biopharmaceutics, Faculty of Pharmaceutical Sciences, The University of British Columbia, Vancouver, British Columbia, Canada
Received, May 7, 2007; Revised, June 1, 2007; Accepted June 22, 2007; Publishes Jul 2, 2007.
Corresponding Author: Kishor M. Wasan, Ph.D. Distinguished University Scholar Professor & Chair Division of Pharmaceutics & Biopharmaceutics Faculty of Pharmaceutical Sciences; The University of British Columbia Vancouver, British Columbia, Canada Telephone: (604) 822-4889; Email address: Kwasan@interchange.ubc.ca
ABSTRACT - PURPOSE. The objective of this study was to determine the influence of two lipid excipients, Peceol® and Gelucire® 44/14 on P-glycoprotein (Pgp) activity and protein expression in human colon adenocarcinoma cells (Caco-2). Lipid excipients are increasingly used as drug delivery systems for hydrophobic drugs to increase their bioavailability by overcoming the barrier of low absorption. .This study will probe a novel mechanism by which lipid excipients reduce Pgp-mediated efflux and thereby increase bioavailability of orally administered therapeutics. METHODS. Non-cytotoxic concentrations of Peceol® and Gelucire® 44/14 were determined for 24-hour treatments of Caco-2 cells using integrity of the cell membranes and mitochondrial respiration as markers. Pgp activity after treatment with non-cytotoxic concentrations of Peceol® and Gelucire® 44/14 was measured with a fluorescent Pgp substrate, rhodamine 123 (Rh123). The activity of Pgp was ascertained by measuring accumulation and the directional flux of Rh123 using the Transwell® semi-permeable cell culture support system. To assess the effect of Peceol® and Gelucire® 44/14 on Pgp protein expression, Western blotting with a specific Pgp antibody was performed. RESULTS. The two assays for cytotoxicity were in agreement and showed that concentrations of less than 0.5% (v/v) Peceol® and less than 0.02% (w/v) Gelucire® 44/14 were not toxic to Caco-2 cells. Rh123 accumulation was increased up to 3-fold in cells treated with sub-toxic concentrations of the excipients. The flux of Rh123 across the cell monolayer was unaffected by treatment in the absorptive (apical to basolateral) direction but the efflux transport was reduced after treatment with Peceol®, Gelucire® 44/14 or the positive control , 100µM verapamil. Some of the reduction in Pgp efflux activity can be explained by the reduction in protein expression after treatment with the lipid excipients; treatment with 0.25% (v/v) and 0.5% (v/v) Peceol® reduced Pgp protein levels to 62.4% and 68.4% of the control respectively while Gelucire® 44/14 treatments of 0.01% (w/v) and 0.02% (w/v) reduced Pgp to 64.5% and 51.8% respectively. CONCLUSION. In this study we utilized established methodologies to assess the inhibitory effect of the excipients on the Pgp-mediated efflux of the probe, Rh123 and tested the hypothesis that long-term treatment of Caco-2 cells with the lipid excipients, Peceol® and Gelucire® 44/14, decreased Pgp protein expression. The results suggest a new mechanism which may contribute to the improved bioavailability seen for drugs formulated with lipid-based excipients.
Introduction
The pivotal activities involved in the development of any oral dosage form include characterization of the active pharmaceutical ingredient (API) and preformulation biopharmaceutics work. The results of these experiments will guide initial excipient selection and a subsequent design of dosage form prototypes. While the activities associated with the development of a lipid-based formulation are similar to those for a conventional oral solid, the manner in which pharmaceutical scientists achieve those goals will be different. The ability of researchers in re-defining these pivotal development activities for lipid-based oral formulations may in fact determine the future success of this technology. The primary roles of traditional excipients are to bind and provide bulk, to the dosage form, to provide a desired drug release profile and to facilitate product manufacturing on high-speed, automated production equipment. Lipid excipients also impart the ability to solubilize hydrophobic drugs within the dosage form matrix. This often results in improved drug absorption, which is primarily mediated by a reduction in the barriers of poor aqueous solubility and slow drug dissolution rate in the GI fluids (1, 2, 3). Some of these excipients also have desirable self-emulsifying properties, readily forming fine dispersions of lipid-solubilized drug in the aqueous contents of the GI tract thereby creating optimal conditions for improved permeability into the enterocytes. Peceol® is a readily dispersible, solubilizing agent comprised primarily of a mixture of mono- and diglycerides of oleic acid, which closely resembles the end-products of intestinal lipid digestion. Gelucire® is a family of naturally derived excipients used in formulations of semi-solid oral dosage forms to improve the solubility of active ingredients in a drug. Gelucire®44/14 combines interesting properties because of its unique composition of surfactants (mono- and diesters of polyethylene glycol, cosurfactants (monoglycerides), and oily phase (di- and triglycerides) (3). The intestinal wall presents the final barrier before a drug enters the body; the primary region responsible for the absorption for orally administered drugs is the jejunum (9). Of the available models to study this system, human colon adenocarcinoma cells, Caco-2 are the best characterized and widely used (9, 26). The interaction of a drug approaching the lipid bilayer is complex, and can be seen as having four distinct regions with distinct hydrophilic and hydrophobic properties (6). Once a drug has penetrated into the cytosol, it is subject to efflux by a variety of transporter proteins; the most significant being P-glycoprotein (Pgp) encoded by the gene ABCB1 (MDR1), multidrug resistance associated protein 2 (MRP2) encoded by the gene ABCC2, and breast cancer resistance protein (BCRP) encoded by the gene ABCG2. Degradation by highly expressed drug metabolizing enzymes of the cytochrome P450 (CYP) and uridine diphosphate–glucuronosyltransferase (UGT) families (5, 9-11) also contributes to poor availability of orally administered medication.
As we begin to unravel the intricacies of the GI processing of lipid excipients, further improvements in the performance of lipid-based delivery systems can be expected. For example, an increasing body of evidence has shown that certain lipids are capable of inhibiting Pgp-mediated drug efflux by the gut wall (12). P-glycoprotein is the best characterized multidrug efflux pump; it is a member of the ATP binding cassette (ABC) superfamily of efflux protein pumps which utilize energy released by hydrolysis of ATP to efflux a variety of xenobiotics (13-15). Pgp-mediated efflux is most noted at the apical face of epithelial cells of the gastrointestinal tract and the blood brain barrier (16, 17).
The mechanism of efflux by Pgp continues to be elucidated and various models have been proposed including (I) the pore model by which the compound is transported out of the cell through a protein channel, (II) the flippase model where the compound is embeded in the inner leaflet of the plasma membrane and then is translocated to the extracellular leaflet through the action of Pgp, and (III) the hydrophobic vacuum cleaner model which combines both the pore and the flippase model (14, 18-20).
Co-administration of drugs with compounds that inhibit Pgp-mediated efflux or the incorporation into specific lipid excipients can alter the pharmacokinetics of the administered compound (20-23). The mechanism by which excipients inhibit Pgp activity is currently unknown, however theories include alteration of the integrity of the cell membrane, blocking binding sites competitively, non-competitively or allosterically, interfering with ATP hydrolysis and creating a futile cycle of ATP hydrolysis (1, 24, 25).
The objective of this study was to determine the influence of two widely used lipid excipients, Peceol® and Gelucire® 44/14, on the transport of a known Pgp substrate, rhodamine-123 (Rh123) and the protein expression of Pgp in a well established cell culture model of intestinal absorption: human colon adenocarcinoma cells, Caco-2 (26).
Materials and METHODS
Materials
Peceol® and Gelucire® 44/14 were a gift from Gattefossé Canada Inc. (Montreal, Quebec, Canada). Verapamil, rhodamine 123 (Rh123), Triton X-100, Tween-80, HEPES, Protease inhibitor cocktail, Na-deoxycholate, EDTA and NaCl were obtained from Sigma-Aldrich (St. Louis, MO, USA). All tissue culture reagents were from Invitrogen/Life Technologies (Grand Island, NY, USA). T-75 flasks and tissue culture treated plates were from Corning Incorporated (Corning, NY, USA). CytoTox96® Non-Radioactive Cytotoxicity Assay and MTS CellTiter 96® AQueous One Solution Cell proliferation Assay were from Promega Corporation (Madison, WI, USA). BCATM Protein Assay Kit was obtained from Pierce Biotechnology, Inc. (Rockford, IL, USA). NP-40 was purchased from Roche Applied Science and Trans-Blot® Transfer medium (nitrocellulose membrane 0.45 mM) from Bio-Rad (Hercules, CA, USA).
Cell Culture
Caco-2, human colon adenocarcinoma cells, were purchased from ATCC (Rockville, MD, USA) and and maintained between passages 20-35. Caco-2 cells were cultured in minimum essential medium with Earl’s salts (EMEM) supplemented with 10% fetal bovine serum (FBS), 292 mg/mL L-Glutamine, 0.1 mM non-essential amino acids, 100 U/mL penicillin and 100 μg/mL streptomycin at 37˚C in humidified air containing 5% CO2. Stock cultures were grown in T-75 flasks and seeded in 96-well, 48-well, 12-well plates, or Transwell® supports depending on the type of experiment. The medium was changed every other day and plates were used for experiments when cells reached 90% confluency.
Cytotoxicity Measurements.
To determine non-cytotoxic concentrations three markers of toxicity were employed: (I) cell plasma membrane integrity as determined by lactate dehydrogenase (LDH) release, (II) mitochondrial respiration as measured by the reduction of a tetrazolium compound (MTS) to a soluble formazan product and (III) total mass of protein recovered from each well as an indication of the number of viable adherent cells. On the day of the experiment, the culture medium was exchanged for Peceol® treatment solutions (0, 0.05, 0.1, 0.25, 0.5, 1.0, 2.0, or 5.0% (v/v)), Gelucire® 44/14 treatment solutions (0, 0.001, 0.002, 0.005, 0.01, 0.02, 0.05, 0.1 or 0.2% (w/v)) or 1% (v/v) Triton X-100 (as positive control for cytotoxicity) in Hanks’ Balanced Salt Solution without phenol (HBSS) containing 10 mM HEPES, pH 7.4. LDH release (CytoTox96® Non-Radioactive Cytotoxicity Assay), MTS reduction (CellTiter 96® AQueous One Solution Cell proliferation Assay) and bicinchoninic acid (BCATM Protein Assay Kit) assays were then performed, and cell viability was calculated relative to the HBSS control (MTS-Assay). Cytotoxicity was calculated relative to the Triton X-100 group (LDH-Assay).
Uptake of Rhodamine-123
For the uptake studies Caco-2 cells were seeded into 48-well plates; on the day of the experiment the culture media was exchanged with fresh culture media or media containing Peceol® (0.1%, 0.25%, 0.50% (v/v)), Gelucire® 44/14 (0.01%, 0.02% (w/v)) or 100µM verapamil for a 24 hour incubation. Cells were washed 3 times with PBS and a solution of fresh HBSS with 10 mM HEPES (pH 7.4) containing 5 mM Rh123 was added to each well. After a 3h incubation the uptake was stopped by aspirating the Rh123/HBSS solution and washing the cells 3 times with ice-cold PBS. Cells were lysed in 1% Triton X-100 and aliquots were used to measure the fluorescence (CytoFluor 4000, Perseptive Biosystems, Framingham, MA, USA; excitation 485 nm and emission 530 nm) and protein content. Cellular accumulation of Rh123 was then normalized with respect to the protein content as determined by BCATM assay in each well.
Rh123 transport studies using Transwell plates
The Caco-2 cells were seeded onto collagen-coated PTFE membrane inserts with a pore size of 0.4 µm and 1.2 cm diameter (Transwell-COL from Corning Costar). The transepithelial electrical resistance (TEER) was monitored using a Millicell-ERS (Millipore, Bedford, MA, USA). Once the TEER value exceeded 300 Ω*cm2 (around 21 days post-seeding) the cell monolayers were used for directional flux experiments. Cells were washed 3 times with PBS before treatment solutions (Peceol: 0.25%, 0.50% (v/v), Gelucire® 44/14: 0.01%, 0.02% (w/v), or 100µM verapamil) were loaded on both the apical side (0.5 mL) and the basolateral side (1.5 mL). After a 24 hour incubation the cells were washed 3 times with PBS and loaded with the appropriate fresh treatment solutions in HBSS (10 mM HEPES) or HBSS (10 mM HEPES) alone. A 5µM Rh123 solution treatment was added to the donor side. Absorptive flux (A-to-B) of Rh123 was measured by removing 50 mL from the basolateral side (replacing with fresh solution to keep the volume constant) after 0, 30, 60 and 120 min. Secretory flux (B-to-A) was measured by taking 50 mL aliquots from the apical side with replacement of treatment solution. The transferred Rh123 concentration was determined by using standard curves of Rh123 in the appropriate treatment solutions. At the end of the experiment the TEER values were measured to assure the integrity of the monolayer.
The apparent permeability coefficient (Papp) of Rh123 was calculated with the following equation:
![]() and
and ![]()
where J0 is the initial flux, C0 the initial Rh123 concentration in the donor compartment at t=0, A is the surface area of the monolayer (in cm2), dQ/dt the drug permeation rate.
The net efflux (Papp ratio) is expressed as the quotient of Papp(B-to-A) to Papp(A-to-B). After the 120min time point, the accumulation of Rh123 in the monolayer was assessed by cutting the membrane and placing it into 1% Triton X-100 to solubilize the cells. The cellular debris and the membrane were pelleted by centrifugation and aliquots from the supernatant were taken for Rh123 content and protein concentration as described above. Rh123 cellular accumulation was then normalized with respect to the protein content in each membrane.
Pgp protein expression: Western blotting
Caco-2 cells were treated for 24 hours with culture medium (control) or culture medium containing Peceol® (0.25%, 0.5% (v/v)) or Gelucire® 44/14 (0.01%, 0.02% (w/v)). Cells were washed 3 times with PBS and harvested with RIPA lysis buffer (50 mM HEPES, 150 mM NaCl, 2 mM EDTA, 0.5% Na-deoxycholate, 1% NP-40) containing protease inhibitor cocktail (1:100 dilution). Protein content was determined using the BCATM assay and the cell membrane proteins (20µg/lane) were separated by electrophoresis through a 10% SDS-polyacrylamide gel at 80V for 120 minutes and then electroblotted at 10V for 90 min onto a nitrocellulose membrane. A pre-stained protein standard was used to identify the Pgp band at 170 kD and actin at 42 kD. The membrane was incubated overnight at 4°C in blocking buffer (1 x PBS, 1% nonfat dried milk, 0.1% Tween-20), and probed for 2h with a 1:300 dilution of mouse anti-human Pgp primary antibody (C219 from Signet Pathology Systems) and a 1:1000 dilution of goat anti-human actin (I-19 from Santa Cruz Biotechnology) to detect actin as an internal control. The membranes were washed 3 times with PBS and 1% Tween-20 (PBS-T) and the membrane was incubated for 90min in a 1:5000 dilution of rabbit anti-mouse IgG horseradish peroxidase (HRP)-conjugated antibody (Jackson ImmunoResearch Laboratories) and a 1:3000 dilution of bovine anti-goat IgG-HRP (Santa Cruz Biotechnology) for Pgp and actin respectively, followed by three washes with PBS-T. Bands were visualized using Western Lightning® Chemiluminescence Reagent (Perkin Elmer Life Sciences), exposed to an X-Ray film (Kodak X-Omat film) and quantified with UVP-Labworks software.
Statistical analysis
All data sets were assessed for normality and equality of variance; one way ANOVA with fixed effects was run followed by Tukey HSD with a predetermined alpha value of 0.05. All statistical analysis was performed using SigmaStat version 3.5 (Systat Inc.).
Results
Measurement of toxicity of Peceol® and Gelucire® 44/14 in Caco-2 cells
Treatment with concentrations 0.05-0.5% (v/v) Peceol® showed no significant difference in cell viability and toxicity compared to the untreated control measured by both MTS (Figure 1A) and LDH assay (Figure 1B). Gelucire® 44/14 is approximately ten times more cytotoxic, the results of the MTS assay depicted in figure 2A show that the cell viability at concentrations up to 0.02% (w/v) are equivalent to the untreated control. Cells treated with 0.05% (w/v) Gelucire® 44/14 showed 85.13 ±32.9 % cell viability relative to control, this difference was not statistically significant but this concentration was not used for future experiments due to the high standard deviations in cell viability. The LDH-assay results (Figure 2B) revealed a similar trend to the MTS assay. These experiments yielded the maximum concentration of Peceol® and Gelucire® 44/14 that could be added to the cell culture system without cytotoxic effects interfering with the interpretation of results. These non-toxic concentrations were subsequently used to treat cells for the Pgp protein expression, Rh123 accumulation and transport studies.
(A)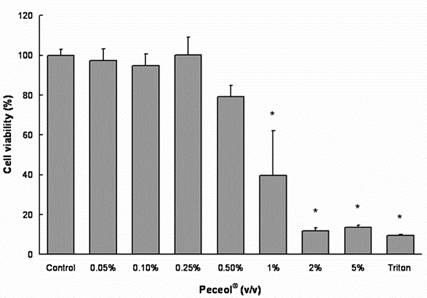
(B) 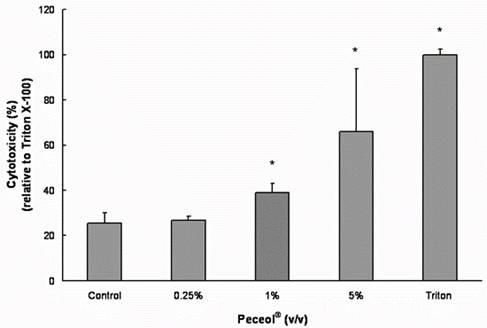
Figure 1. Effects of Peceol® on Cell viability and cytotoxicity in Caco-2 cells. (A) MTS assays were performed to measure the survival rate of Caco-2 cells after treatment with Peceol®. Data are expressed by the mean of percent cell viability compared to control after exposure for 24 hours ± standard deviation (n=4-6). (B) Cytotoxicity was measured by LDH-assays and compared to Triton X-100. Each bar represents the mean ± standard deviation of four to six experiments (* P<0.05).
(A)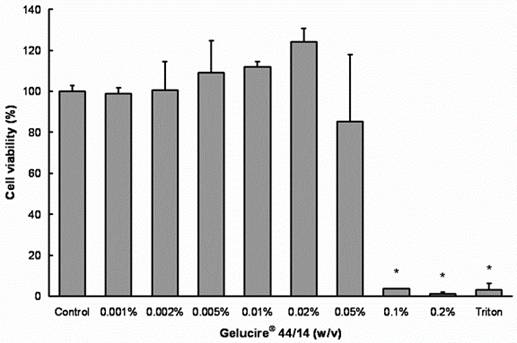
(B)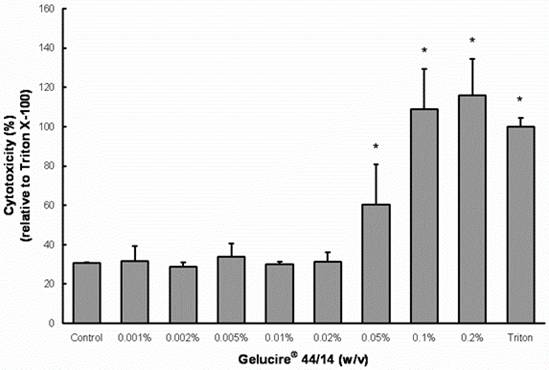
Figure 2. Effects of Gelucire® 44/14 on Cell viability and cytotoxicity in Caco-2 cells.
(A) MTS assays were performed to measure the survival rate of Caco-2 cells after treatment with Gelucire® 44/14. Data are expressed by the mean of percent cell viability compared to control after exposure for 24 hours ± standard deviation (n=4). (B) Cytotoxicity was measured by LDH-assays and compared to Triton X-100. Each bar represents the mean ± standard deviation of four to six experiments (* P<0.05).
Role of Peceol® and Gelucire® 44/14 in enhanced accumulation of Rh123
All treatments with Peceol® and Gelucire® 44/14 resulted in a significant increase in Rh123 accumulation relative to the untreated control cells (Figure 3: p<0.05). The Rh123 accumulation in the treatment groups did not differ significantly from the accumulation of Rh123 in the verapamil-treated cells.
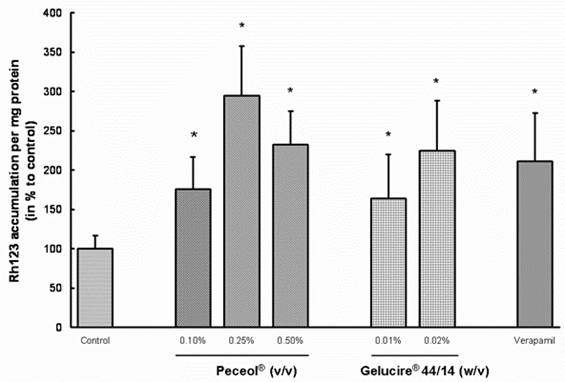
Figure 3. Peceol® and Gelucire® 44/14 enhance the accumulation of Rh123 in Caco-2 cells.
Caco-2 cells were pretreated for 24 hours with 0.1, 0.25 and 0.5 % (v/v) Peceol® ,0.01 and 0.02% (w/v) Gelucire® 44/14 or 100 mM verapamil as control for Pgp inhibition. Cells were washed and fresh treatment solutions plus 5 mM Rh123 were added for another 3 hours and the accumulation of Rh123 in lysed cells was determined. Rh123 accumulation in Peceol®, Gelucire® 44/14 and verapamil treated Caco-2 cells was compared to untreated control cells. Data expressed as the mean percent to control normalized for the protein ± standard deviation for n=6 (*P<0.05).
Effect of Peceol® and Gelucire®44/14 on the transport of Rh123 across Caco-2 cells
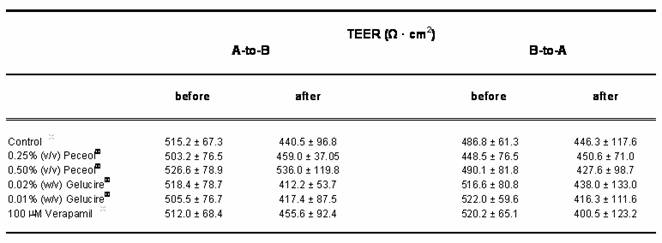
The flux of Rh123 across a Caco-2 monolayer in both the absorptive (apical to basolateral) and secretory (basolateral to apical) directions was assessed after treatment with Peceol® (0.25%, 0.5% (v/v)), Gelucire® 44/14 (0.01%, 0.02% (w/v)) or 100µM verapamil; table one summarizes the results of these experiments. All treatments showed a statistically significant decrease in secretory flux compared with the untreated control cells (p<0.05); however the differences in absorptive flux were not found to be statistically significant. TEER values were obtained prior to and after each experiment and no significant differences were noted (Table II).
Effects of Peceol® and Gelucire®44/14 on P-glycoprotein expression in Caco-2 cells
P-glycoprotein expression was measured in Caco-2 cells which had been treated for 24 hours with Peceol®, Gelucire®44/14 or control media. Peceol® significantly decreased the expression of Pgp for the two tested Peceol® concentrations compared to control (Figure 4) to 62.4 ± 21% for 0.25% (v/v) and to 68.4 ± 20% for 0.5% (v/v) Peceol®.
Treatment with Gelucire®44/14 also lead to a significant decrease in Pgp expression (Figure 5). The Pgp expression after a 24h treatment with 0.01% and 0.02% (w/v) Gelucire® 44/14 was significant lower than the corresponding mean values determined for control cells, 64.5 ± 28 % and 51.8 ± 32% respectively.
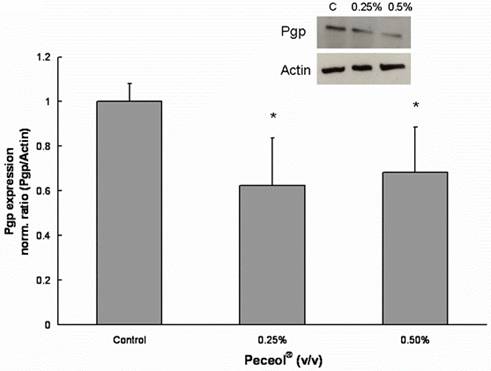
Figure 4. Peceol® inhibits the Pgp protein expression in Caco-2 cells.
Pgp protein expression after 24 hour exposure to Peceol®. 20 mg of total protein were separated by SDS-polyacrylamide gel electrophoresis and immunoblotted with monoclonal antibody C219 for Pgp and I-19 for actin as described under Materials and Methods. The insert is an image of a representative blot. Each bar represents the mean ± standard deviation of Pgp protein expression normalized by actin expression. Pgp expression in treatment groups were compared with Pgp expression in untreated control cells. n=4 (*P<0.05).
Discussion
In this study we evaluated the activity and expression of P-glycoprotein in a human intestinal cell line in the presence and absence of two lipid excipients: Peceol® and Gelucire®44/14. Both excipients are commonly used in the pharmaceutical industry to increase bioavailability of hydrophobic drugs.
The primary mechanism by which lipid excipients improve the bioavailability of orally administered therapeutics is through improved dissolution rates in the GI environment. Gelucire® 44/14 has a hydrophilic/lipophilic balance of 14 indicating a hydrophilic nature yet is suitable for solubilizing even poorly soluble molecules (20, 35, 36). Lipid excipients have the potential to be more than simply a drug delivery system; the results of this study indicate that there are interactions between the lipid-based formulations and the gastrointestinal system. There is a growing body of evidence that certain lipids can inhibit presystemic drug metabolism and can play an active role in Pgp mediated efflux from the enterocytes (27, 28). The findings of this study expand on a previous publication in which we demonstrated that Peceol® down regulated MDR1 gene expression and Pgp protein expression (29) in the Caco-2 cell culture system. Although Pgp is a well characterized transporter, many studies have focussed on drug-protein interactions and little is known about the effects of lipid excipients. In a recent review article, Constantinides and Wasan summarize the state of knowledge concerning lipid formulations (12) however it remains unclear if and how transporter expression is influenced by formulation components assumed to be inert.
Using established methodologies, such as accumulation and directional transport, we tested whether Peceol® and Gelucire® 44/14 influenced the efflux of a known Pgp substrate, Rh123. The experiments were conducted at concentrations of excipient tested to be below cytotoxic levels to ensure that the effects were not a by-product of toxicity-induced reactions. Verapamil was selected as the positive control for inhibition of Pgp activity due to the frequency of citations which make use of its inhibitory properties. The selection of an appropriate positive control for Pgp inhibition can be a daunting task. The mechanisms of inhibition proposed for pharmaceutical excipients have focused on the disruption of membrane fluidity and reduction in the affinity of ATP for Pgp. First generation chemical inhibitors, including verapamil, quinidine and cyclosporine A were developed for other indications and tend to lack potency and selectivity for Pgp (3, 30-33). Newer compounds such as PSC 833 and GF120918 show improved specificity and potency (3, 5, 34) but may not be the optimal controls when examining excipient interactions. Additionally, the mechanisms by which all of these compounds inhibit Pgp activity are through competitive inhibition for the drug binding domains of the protein.
In our Rh123 accumulation studies, the highest intracellular concentration of Rh123 was observed in 0.25% and 0.5% (v/v) Peceol® and 0.02% (w/v) Gelucire® 44/14 treated cells (Figure 3). The levels exceeded even the concentrations detected in the verapamil treated cells. The 0.1% (v/v) Peceol® and 0.01% (w/v) Gelucire® 44/14 treatment groups also showed significant increases relative to the media control. Directional flux of Rh123 across the cell monolayer was selected as a second assay for Pgp activity; both Peceol® and Gelucire® 44/14 treatment suppressed the secretion of Rh123. This reduction in the secretory pathway was caused by a statistically significant reduction in the basolateral to apical flux; the apical-to-basolateral flux did not show significant changes (Table I). The unchanged TEER values (Table II) and total cellular protein content indicated that the reduced activity of the transporter was not caused by cell damage or cell death.
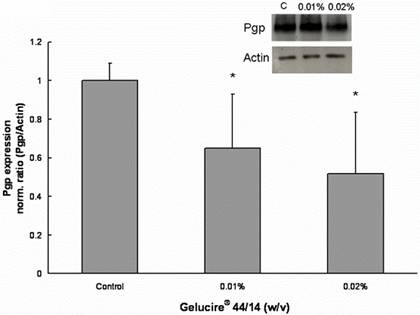
Figure 5. Gelucire® 44/14 inhibits the Pgp protein expression in Caco-2 cells. Pgp protein expression after 24 hour exposure to Gelucire® 44/14. 20 mg of total protein were separated by SDS-polyacrylamide gel electrophoresis and immunoblotted with monoclonal antibody C219 for Pgp and I-19 for actin as described under Materials and Methods. The insert is an image of a representative blot. Each bar represents the mean ± standard deviation of Pgp protein expression normalized by actin expression. Pgp expression in treatment groups were compared with Pgp expression in untreated control cells. n=4 (*P<0.05).
Table I. Apparent Permeability Coefficients (Papp) for apical to basolateral and basolateral to apical transfer of Rh123 across Caco-2 cell monolayer.
The apparent permeability coefficient (Papp) of rhodamine 123 was calculated with the following equation:
![]() and
and ![]()
where J0 is the initial flux, C0 the initial Rh123 concentration in the donor compartment at t=0, A is the surface area of the monolayer (in cm2), dQ/dt the drug permeation rate. The net efflux (Papp ratio) is expressed as the quotient of Papp(B→A) to Papp(A→B). Data expressed as mean ± standard deviation for n=4 to 6 compared control (*P<0.05).
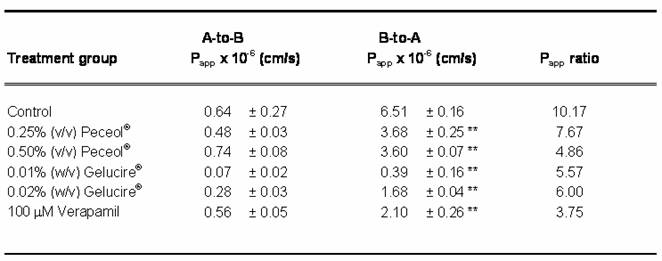
Table II. The effect of Peceol®, Gelucire® 44/14 and Verapamil on the Transepithelial Electrical Resistance (TEER). TEER values of Caco-2 cell monolayer were measured before and after the transport of Rh123 in the apical-to-basolateral and basolateral-to-apical direction in the presence of 0.25% and 0.5% (v/v) Peceol®, 0.01% 0.02% (w/v) Gelucire® 44/14 and 100 mM verapamil. TEER ratio = TEERafter / TEERbefore Data presented as mean ± standard deviation with n=3-6.
The Rh123 transport profiles obtained for Peceol® and Gelucire® 44/14 are similar to the profile produced by treatment with 100 mM verapamil. The two excipients were as efficient in abolishing the vectorial transport of Rh123, as measured by a significant decrease in the basolateral to apical apparent permeability (Papp) accompanied by no changes in apical to basolateral apparent permeability (Table I). Comparison of the Papp ratios for Peceol® and for Gelucire®44/14 with the Papp ratio obtained for untreated controls indicated that active secretion by the efflux transporter was significantly reduced in the treatment groups. The pivotal role of Pgp in the transport of Rh123 leads us to conclude that Peceol® and Gelucire® 44/14 achieved the reductions in Rh123 secretion via inhibition of Pgp activity.
Several mechanisms by which excipients nonspecifically inhibit Pgp activity have been proposed and carefully studied. Surfactants and solvents have been shown to modulate Pgp activity through altered fluidity of the lipid membrane environment of Pgp leading to a reduction of ATPase activity (37, 38). Batrakova et al. showed that pluronic block copolymers sensitize multridrug resistant cell lines by decreasing the affinity of Pgp for ATP thereby decreasing the ATPase activity in combination with depletion of intracellular ATP (21). Zastre et al. (39) describe a model for Pgp inhibition in which methoxypolyethylene glycol-block-polycaprolactone (MePEG-b-PCL) copolymers enhanced Rh123 accumulation in intestinal cells and decreased the Papp ratio (B-to-A)/(A-to-B). With MePEG-b-PCL, Pgp inhibition only occurred at concentrations above the critical micelle concentration (CMC); other groups have shown Pgp inhibition by other surfactants at concentrations below the CMC (1, 40, 41).
Our decreased Pgp protein expression data in Caco-2 cells after treatment with Peceol® and Gelucire® 44/14 and our earlier findings of downregulation of MDR1 (29) suggest that more than just changes in membrane fluidity or ATPase activity are responsible for lower Pgp activity.
Studies focusing on MDR1 have shown that elevated expression can be induced by carcinogens, chemotherapeutic substances, UV radiation, inflammation , heat shock and heavy metals (42-45). Recently, transcriptional control of ABC transporters has been linked to a group of nuclear receptors implicated in a hierarchical network of transcriptional control for a variety of processes including nutrient, bile acid, and xenobiotic metabolism and other metabolic functions (46). Many nuclear receptors have lipid ligands providing interplay between lipids and transcriptional control of ABC transporters (47-50). The nuclear receptor proteins retinoic acid receptor (RAR), farnesoid receptor (FXR), steroid-activated receptor (SXR) and pregnane receptor in rodents (PXR) have previously been identified in the transcriptional control of transporter proteins (51, 52), PXR in particular has been implicated in xenobiotic protection (46). Future studies with lipid-derived excipients will be focused on identification of the receptor pathways related to the observed decreases in P-glycoprotein expression.
In summary we can conclude from our findings that lipid excipients can play an active role in affecting Pgp mediated transport not only by altering the membrane fluidity or ATPase activity but by also downregulating P-glycoprotein expression. If lipid excipients indeed influence transporter expression, this needs to be recognized by pharmaceutical and formulation scientists as well as regulatory agencies.