J Pharm Pharmaceut Sci (www.cspscanada.org) 10(3):311-318, 2007
Multiple oral dosing of ketoconazole increases dog exposure to ivermectin
Christophe Hugnet, Anne Lespine, Michel Alvinerie
Clinique Vétérinaire des Lavandes, 26160 La Begude de Mazenc, France. INRA-UR66, Laboratoire de Pharmacologie-Toxicologie, BP 3, 31931 Toulouse Cedex 9, France
Received, January 29, 2007; Revised, May 18, 2007; Accepted, May 29, 2007, Published, June 23, 2007
Corresponding Author: Michel Alvinerie, INRA-Toulouse, Laboratoire de Pharmacologie-Toxicologie, BP 3, 31931 Toulouse Cedex 9, France Phone : 33-561285137, E-mail : michel.alvinerie@toulouse.inra.fr
ABSTRACT - Purpose. The parasiticide ivermectin and the antimicrobial drug ketoconazole are macrolides that interact with P-glycoprotein. We investigated the effects of ketoconazole at a clinical dose on the pharmacokinetics of ivermectin, a CYP3A substrate with low hepatic clearance. Methods. Beagle dogs received a single subcutaneous injection of ivermectin at 0.05 mg/kg alone (n=6) or in combination with a daily oral dose of ketoconazole 10 mg/kg over 5 days before and after ivermectin administration (n=6). The plasma kinetics of ivermectin and its metabolite were followed over 15 days by HPLC analysis. Results. Co-administered ketoconazole induced a higher plasma concentration and longer residence time of ivermectin in dogs, leading to a substantial increase in the overall exposure of the animal to the drug. Ketoconazole does not interfere with the production of the ivermectin metabolite but it may rather inhibit the elimination of the parental drug by interfering with P-gp transport. Conclusions. Multiple oral dosing of ketoconazole dramatically altered the pharmacokinetics of ivermectin in dogs leading to an increase in systemic exposure to the drug. Neurotoxicity of ivermectin means that inhibition of the P-gp function at the blood-brain barrier during polytherapy using P-gp inhibitors must be taken into consideration.
Introduction
The macrocyclic lactones (MLs) belong to a large family of structurally related compounds widely used in humans and animals as a potent endo- and ectoparasiticides (1). In dogs, they are used in the prevention of dirofilariosis, as roundworm worming and in the treatment of some ectoparasitosis such as mange and demodecosis. In farm animals, they are the most frequently used molecules worldwide for their wormer properties as well as for their ectoparasiticide properties. In human medicine, ivermectin is mainly prescribed in tropical and subtropical regions for the treatment of onchocercosis (Onchocerca volvulus), some filariosis (Wuchereria bancrofti) as well as sarcoptic mange (2, 3). Many other claims have been made such as the treatment of demodicosis, larva migrans syndromes, myiasis (4, 5). With the emergence of human immunodeficiency virus (HIV) in the past three decades, ivermectin is also used in Western countries for the treatment of opportunistic parasitic diseases.
It has been clearly demonstrated that the ML ivermectin and structurally related compounds interact with P-glycoprotein (P-gp) (6-8). P-gp is a 170 kDa transmembrane protein that belongs to the ATP binding cassette (ABC) protein superfamily (9) and which is coded for by the MDR1 gene (ABCB1). P-gp is involved in the active cellular efflux of a large number of drugs and toxic compounds and transports many structurally and pharmacologically unrelated hydrophobic compounds including anticancer agents, immunosuppressive agents, steroid hormones, calcium channel blockers, β-blockers, pesticides, anthelmintics, antibiotics, HIV protease inhibitors and cardiac glycosides (10). Most of these P-gp substrates are also substrates of the major drug-metabolizing 3A4 isotype of the cytochrome P450 enzymes. In the central nervous system (CNS), P-glycoprotein is found in the capillary-endothelial cells that form the blood-brain barrier. The avermectins are transported by P-glycoprotein from the inside to the outside of the endothelial cells back into the lumen of the capillary, thus preventing further diffusion in the CNS. In the absence of P-glycoprotein, the avermectins are capable of diffusing freely and accumulating into the CNS, leading to neurotoxicity of the drug. Indeed, P-gp deficient animals such as a subpopulation of the CF-1 mouse strain (11) or genetically engineered mice (8) as well as certain dogs of the Collie breed (12-15), are unusually sensitive to the adverse effects of ivermectin.
In humans, rare cases of intolerance to ivermectin leading to neurological symptoms have also been reported in Cameroon. The cause has not yet been identified with certainty, but a defect in MDR1 or a P-gp interaction with drugs or foodstuffs are suspected (16-18).
Ketoconazole, an imidazole derivative, is an orally active, broad spectrum systemic antifungal agent extensively used in veterinary medicine. Ketoconazole is especially effective against fungal diseases in dogs including dermatophytosis (19), blastomycosis (20) aspergillosis (21) and cryptococcosis (22), and hyperadrenocorticism (23).
Ketoconazole is well-known to potently inhibit CYP3A4 activity in human liver microsomes (24, 25) and modifies considerably the pharmacokinetics of CYP3A4 substrates such as cyclosporine (26) and tacrolimus (27). Furthermore, ketoconazole competitively inhibits CYP3A12, a major isoenzyme of the CYP3A subfamily in canine hepatic microsomes (28), and at a therapeutic dose, decreases the total body clearance of the CYP3A substrates midazolam (29) and nifedipine (30) in beagle dogs. Therefore, in clinical practice, adverse drug effects may develop when ketoconazole is administered concomitantly with drugs that are primarily metabolized by CYP3A. Ketoconazole also inhibits P-gp function in the monkey (31).
In this study, we investigated the effects of multiple oral dosing of ketoconazole at a clinical dose, on the pharmacokinetics of ivermectin, a CYP3A substrate with low hepatic clearance, when subcutaneously administered to beagle dogs.
Materials and METHODS
Materials
Ketoconazole was purchased as tablets (Ketofungol®) from Janssen Pharmaceutica (Issy les Moulineaux, France). Ivermectin injectable solution (Ivomec®, 1%) was obtained from Merial (Lyon, France). All other chemicals used as reagents were of analytical and high-performance liquid chromatographic (HPLC) grade. The standard for the ivermectin metabolite, the 3 O-desmethyl ivermectin, was kindly provided by Merial (Iselin, USA).
Animals
Twelve male beagle dogs (1 to 7 year old) weighing 13–18 kg were bought from a breeder. The dogs were individually housed and allowed access to water ad libitum and were given dry food twice a day (8:00 a.m. and 8:00 p.m.). They were vaccinated and dewormed according to the standard guidelines (latest boost and wormer: 3 months previous to the study). No other treatment had been given administered during the 90 days prior to the study. All procedures adhered to the “Guide to the Care and Use of Experimental Animal Care” (Canadian Council on Animal Care guidelines, 1984) and the protocol was approved by the local animal ethics committee.
Study design
The animals were randomly allocated into two groups of six animals. One group received ivermectin alone (Ivm group) and the other group received ivermectin plus ketoconazole (KTZ-Ivm group). Ivermectin was administered to the dogs subcutaneously at 0.05 mg.kg-1. This dose, which corresponded to a quarter of usual dose, was chosen because ivermectin neurotoxicity was suspected in ivermectin insensitive breeds of dogs given ivermectin (0.2 mg.kg-1) with ketoconazole (personal communications from Veterinary Pharmacovigilancy Center of Lyon). Dogs from the KTZ-Ivm group received the same ivermectin dose plus 10 mg/kg of ketoconazole orally (1 hr after each morning meal) over 10 days (5 days before and 5 days after the ivermectin injection).
Blood sampling and determination of plasma ivermectin concentration
Blood samples (1 ml) were collected from the cephalic vein before the ivermectin injection and at 1, 2, 4, 8, 24, 36 h, and 2, 3, 4, 6, 8 and 11 days post administration. Plasmas were immediately separated from whole blood by centrifugation for 10 min at 1500 x g and stored at −18 °C prior to analysis.
Ivermectin was analyzed in plasma by high-performance liquid chromatography (HPLC) with automated solid phase extraction and fluorescence detection according to a previously described method (32). Briefly, plasma samples were diluted in acetonitrile, mixed for 20 min, sonicated for 10 min (Ultrasound Bath) and centrifuged at 2000 × g for 2 min. The supernatant obtained was submitted to an automated phase extraction and placed on a Benchmate apparatus (BenchMate II, Zymark, Hopkinton, MA, USA). The fluorescent derivative was obtained by dissolving the eluent in N-methylimidazole and trifluoroacetic anhydride (Aldrich, Milwaukee, WI, USA) solutions in acetonitrile. The chromatographic conditions consisted of a mobile phase of 2% acetic acid, methanol, acetonitrile (4:32:64, v/v/v) pumped at a flow rate of 1.5 ml.min-1 through a Supelcosil C18, 3 μm column (150 mm × 4.6 mm) (Supelco, Bellefonte, PA, USA). Fluorescence detection (Detector RF 551, Shimadu, Kyoto, Japan) was performed at 365 nm excitation and 475 nm emission wavelengths.
Pharmacokinetic analysis
Data were analyzed using a compartmental approach with the KineticaTM (version 4.2) computer program (InnaPhase, Philadelphia, USA). The following bi-exponential equation was fitted to the plasma concentration vs. time data using a program for non-linear progression analysis adapted from MULTI, based on Akaike’s information criterion (33):
C(t) = A1e-t – A2e-kat
In this equation, A1 and A2 are intercepts, C(t) is the plasma concentration at time t, ka is the estimated first order rate constant of ivermectin absorption, and is the first order rate constant of ivermectin elimination. The data were also subject to non-compartmental analysis using the statistical moment approach (34).
The terminal (elimination) half-life (t1/2el), and absorption half-life (t1/2) were calculated as ln2/ka, and ln2/ka, respectively. The areas under the plasma concentration-time curves (AUC) from time zero to the last time with a measurable concentration were calculated using the linear trapezoidal rule. The mean residence time (MRT) was calculated using the linear trapezoidal rule without extrapolation to infinity, using the formula:
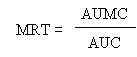
where AUMC is the area under the momentum curve and AUC the area under the plasma concentration time curves, as previously defined. The peak concentration (Cmax) and time to reach peak concentration (Tmax) were read from the plotted concentration-time curves for each animal.
Statistical analysis
The mean pharmacokinetic parameters for the two groups were statistically compared with unpaired t-test analysis using the Statview statistics program (version 5.0). Mean values were considered significantly different at p < 0.05.
Results
No adverse effects of the treatment were observed in the two groups throughout the experimental period. The analytical method used to extract, derivatize and quantify the plasma concentration of ivermectin by chromatographic analysis using fluorescence detection was validated. Calibration graphs in the range 0.1-50 ng/ml-1 were prepared using pooled drug free plasma and taken through the procedure. The regression line of the plot of peak area against drug concentration showed a correlation coefficient of 0.995. The mean recovery from the extraction from plasma was 95 %. The interassay precision showed a variation coefficient of 4.7 % and the limit of quantification was established as 0.1 ng.ml-1. The graph of the mean plasma concentration vs. time of ivermectin after subcutaneous administration in the two groups is shown in Fig.1. In both groups, ivermectin was detected in plasma between 1 hour and 11 days after administration. Pharmacokinetic parameters of ivermectin obtained in standard dogs were in full agreement with those reported in a previous study (35). Concomitant administration of ketoconazole induced substantial increases in plasma concentrations of ivermectin resulting in a significant increase in the AUC (2-fold, p<0.001). Data for the main pharmacokinetic parameters describing the disposition of ivermectin in both groups are shown in Table 1. The mean MRT was significantly increased from 3.8 ±0.8 days in the Ivm group to 5.8 ±1.1 days in the KTZ-Ivm group (p<0.006). In the latter, the half-life of elimination (t1/2) was also increased, but without reaching statistical significance.

Figure 1. Ivermectin concentration vs. time curves in dog plasma following a subcutaneously administered dose of 0.05 mg.kg-1 without (--o--) or added with ketoconazole (-■-). Data are the mean ± SE of six animals in each group.
At the same time, the mean Kel and Cl/F were significantly decreased (by 1.7- and 2.2-fold, respectively) by drug co-administration. A metabolite of ivermectin was detected and quantified in the dogs’ plasma. By comparison with previous studies in goats and pigs, this compound was identified as being the 3 O-desmethyl ivermectin (36, 37). The AUC of ivermectin and the metabolite in both groups are shown in Fig.2. In our study, this metabolite accounted for 11.4 % of the ivermectin AUC in the Ivm group and 9.2 % of the ivermectin AUC in the KTZ-Ivm group, and its concentration vs. time profiles closely paralleled those of ivermectin in both groups. Although in the KTZ-Ivm animals the metabolite concentration was higher, the ratio of ivermectin/metabolite remained constant throughout the kinetics and was similar in both groups. Similarly, the ratios of the AUCs of the metabolite vs. the parent ivermectin were quite similar in both groups with 0.108 ±0.026 ng.d.ml-1 for the Ivm group and 0.092 ±0.017 ng.d.ml-1 for the KTZ-Ivm group.
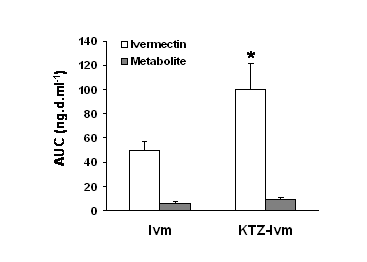
Figure 2. Effect of ketoconazole on the ivermectin or metabolite area of the plasma concentration vs. time (AUC) in the Ivm group and KTZ-Ivm group. Data are the mean ± SE of 6 animals in each group. * significantly different when compared with the Ivm group, p<0.05.
Discussion
The purpose of this study was to investigate the effect of multiple oral dosing of ketoconazole at therapeutic doses on the pharmacokinetics of ivermectin in dogs. The results clearly demonstrated that co-administered ketoconazole induced higher plasma concentrations and a longer residence time of ivermectin in dogs, leading to a substantial increase in the overall exposure of the animal to the antiparasitic drug. In the ketoconazole co-administered group, there was a good concordance between the increase AUC and MRT that are parameters of exposure to ivermectin and the observed decrease in the parameters of elimination (Cl/F and Kel). Indeed, the clearance and constant of elimination of ivermectin were significantly lower in the group receiving the drug combination. Altogether, the increased plasma levels of ivermectin and the reduced elimination strongly suggest that ketoconazole increased the exposure of the animals to the drug by decreasing the elimination process.
Since ketoconazole is known to inhibit both CYP3A and the drug efflux transporter P-glycoprotein, both pathways might contribute to the increase in the exposure of the dogs to ivermectin. In fact, there are reports that the co-administration of ketoconazole increases the bioavailability of highly metabolized drugs such as quinidine (38), mefloquine (39), fexofenadine (40), quetiapine (41) and everolimus (42) in humans.
Due to the inhibitory effect of ketoconazole on CYP3A, one might expect that the administration of ketoconazole would decrease the production of the ivermectin metabolite, thus explaining the increase in the concentration of ivermectin in the systemic circulation. In the present experiment, a main metabolite was identified as the 3 O-desmethyl ivermectin, which has been previously reported in goats (36) and pigs (37) but never explored in dog. The concentration of this metabolite was low in both experimental groups never exceeding 12 % of the total ivermectin in plasma, confirming that ivermectin is a poorly metabolized drug as previously shown in several animal species (37). Moreover, the ratios of parental ivermectin to metabolite concentrations in plasma were constant throughout the experimental period. These results clearly demonstrate that the increase of exposure to ivermectin observed in animals receiving both ivermectin and ketoconazole was not due to the inhibition of ivermectin metabolism by ketoconazole.
Concerning the drug efflux, it is clearly demonstrated that the plasma and tissue distribution of ivermectin was strongly influenced by P-gp activity. The strategic distribution of P-gp on the biliary canicular membrane of hepatocytes and on the apical side of enterocytes means that this ABC-transporter is a key player in both the biliary and the intestinal secretion of ivermectin. Indeed, the disposition of ivermectin in the intestinal fluid and tissue was significantly modified in the presence of P-gp modulators under both in-vitro and in-vivo conditions (43, 44). Subsequently, enhanced ivermectin bioavailabilty was observed in-vivo by the concurrent administration of ivermectin with verapamil (45, 46), a well-known competitive substrate for the P-gp drug-binding site and with itraconazole (43) or loperamide (47). In addition, ketoconazole co-administration increased the AUC of several P-pg substrate drugs in rats (48).
On the basis of our results, we suggest a mechanistic explanation for the decreased ivermectin elimination in dogs treated with ketoconazole: ketoconazole does not interfere with the production of the ivermectin metabolite, but rather inhibits the elimination of the parental drug certainly through its potential to interfere with P-gp transport function. Because ketoconazole certainly inhibits biliary and intestinal P-gp-mediated excretion of ivermectin, it decreases the clearance of ivermectin and increases the drug concentration in the organism.
Because of its location on the blood-brain barrier, the role of P-gp is particularly important for the protection against ivermectin accumulation in the central nervous system and its subsequent neurotoxicity (8, 13, 14). The ketoconazole-ivermectin interaction described in the present study may lead to an unexpected high level of systemic ivermectin that represents a major concern for animals or humans with altered P-gp function.
In addition to physiological and environmental factors or therapeutics, the expression and function of P-gp are modified by genetic polymorphisms of the MDR1 gene (49). However, in contrast to drug-metabolizing enzymes for which mutations leading to loss of function or gene amplification manifest as distinct phenotypes in the population, the impact of MDR1 polymorphisms on pharmacokinetics and pharmacodynamics of P-gp substrates is moderate (50). By contrast to the dog, on the basis of its wide use in man over 20 years, ivermectin appears to be safe in humans. However, suspicion of ivermectin neurotoxicity related to an alteration in P-gp expression has been reported in a subpopulation living in Cameroon (16-18). Because P-gp can be simultaneously targeted by multiple physiological and environmental factors and by therapeutics, one cannot rule out that a hidden ivermectin P-gp sensitivity could be revealed, for example during poly-therapy, particularly in poly-parasitized patients and/or in immunodepressed humans treated with HIV protease inhibitors.
These data indicate that a greater understanding of P-gp drug-drug interactions is needed and that lower doses of ivermectin should be given when it is used in association with another P-gp interfering drug, in order to avoid neurotoxic adverse effects. Clinicians should be aware of such interactions and caution must be exercised in veterinary and human clinical practice, where both ketoconazole and ivermectin are widely prescribed.
Table 1. Pharmacokinetic parameters of ivermectin obtained in the two groups of dogs after a single subcutaneous administration of 0.05 mg.kg-1.
Parameters |
Ivermectin |
Ivermectin + ketoconazole |
ANOVA |
AUC (ng.days.ml-1) |
50.13 ± 7.01 |
104.81 ± 21.31 |
0.001 |
Cmax (ng.ml-1) |
11.59 ± 3.02 |
15.92 ± 4.78 |
0.089 |
Tmax (days) |
1.42 ± 0.72 |
2.46 ± 1.06 |
0.073 |
MRT (days) |
3.84 ± 0.81 |
5.76 ± 1.11 |
0.006 |
t1/2 β (days) |
2.12 ± 0.20 |
2.85 ± 0.51 |
0.085 |
K (per day) |
1.93 ± 1.54 |
0.95 ± 0.66 |
0.181 |
Kel (per day) |
0.35 ± 0.05 |
0.21 ± 0.05 |
0.001 |
Clearance (m/day/kg) |
974±136 |
440 ± 103 |
0.001 |
Cmax, observed peak plasma concentration ; Tmax, time to reach Cmax ; t1/2 β, half-life of elimination ; MRT, mean residence time; AUC, area under the plasma concentration vs. time curve; ka, constant rate of absorption ; kel, constant of elimination. Values are mean ± SD and are considered as significantly different when p < 0.05.
CONCLUSIONS
Multiple oral dosing of ketoconazole dramatically altered the pharmacokinetics of ivermectin in dogs leading to an increase in systemic concentrations of the drug. Too high levels of ivermectin could be toxic to ivermectin-sensitive animals where such drug association must be avoided. In order to optimise antiparasitic drug usage, on the basis of our data, lowering the therapeutic dosage of ivermectin should be envisaged when it is co-administered with ketoconazole.