J Pharm Pharmaceut Sci (www.cspscanada.org) 10(1):37-50, 2007
Dose-dependent pharmacokinetics of telithromycin after intravenous and oral administration to rats: Contribution of intestinal first-pass effect to low bioavailability
Joo Hyun Lee, Myung Gull Lee
College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, Seoul, South Korea.
Received, December 12, 2006; Revised, March 10, 2007; Accepted, Match 20, 2007, Publishes, March 21, 2007.
Corresponding Author: Myung G. Lee, College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, South Korea, leemg@snu.ac.kr
ABSTRACT -- Purpose. To evaluate the pharmacokinetics of telithromycin after intravenous and oral administration and to find the reason for incomplete F value (first pass-effect) after intravenous, intraportal, intragastric, and intraduodenal administration to rats. Methods. Telithromycin was administered intravenously or orally at doses of 20, 50, and 100 mg/kg to rats. And hepatic, gastric, and intestinal first-pass effects of telithromycin were also measured after intravenous, intraportal, intragastric, and intraduodenal administration at a dose of 50 mg/kg to rats. Results. The dose-normalized AUC values of telithromycin were dose-dependent (increased with increasing doses) after both intravenous and oral dose ranges studied, possibly due to saturable metabolism of telithromycin. After oral administration (50 mg/kg), approximately 4.06% of oral dose was not absorbed, F was approximately 27.5%, and the intestinal first-pass effect was approximately 63.4% of oral dose. The first-pass effects of telithromycin in the lung, heart, stomach, and liver were almost negligible, if any, in rats. Conclusions. The low F of telithromycin at a dose of 50 mg/kg was mainly due to considerable intestinal first-pass effect, approximately 63.4% of oral dose, in rats.
Introduction
Telithromycin (Figure 1), a ketolide, is the first of a new class of semisynthetic agents derived from erythromycin. It inhibits bacterial protein synthesis via the two mechanisms; first by directly blocking translation of mRNA and second by interfering with the assembly of new ribosomal units (1). Telithromycin has been developed for the treatment of upper and lower community-acquired respiratory tract infections. Telithromycin has potent activities both in vitro and in vivo against common respiratory tract pathogens including Streptococcus pneumoniae, Haemophilus influenza, Moraxella catarrhalis, and Group A β-haemolytic streptococci, irrespective of their β-lactam or macrolide susceptibility (2). Its spectrum of activity also extends to atypical and intracellular pathogens (3). The total body clearance, volume of distribution, half-life, plasma protein binding, urinary excretion, and extent of absolute oral bioavailability (F) of telithromycin in humans were 14 mL/min/kg, 3.0 L/kg, 12 h, 70%, 23%, and 57%, respectively (4–6). Although the pharmacokinetic studies on telithromycin in humans have been reported (4–7), the reason for the incomplete F of the drug did not seem to be reported. Hence, the present study was performed.
The purpose of this study is to report the dose-dependent total area under the plasma concentration–time curve from time zero to time infinity (AUC) of telithromycin after intravenous (at doses of 20, 50, and 100 mg/kg) and oral (at doses of 20, 50, and 100 mg/kg) administration of the drug to rats, and the first-pass (hepatic, gastric, and intestinal) effects of telithromycin after intravenous, intraportal, intragastric, and intraduodenal administration of the drug at a dose of 50 mg/kg to rats.
Materials and METHODS
Chemicals
Telithromycin was kindly supplied from Aventis Pharma Deutschland GmbH (Frankfurt am Main, Germany). Quinine hydrochloride [an internal standard of high-performance liquid chromatographic (HPLC) analysis of telithromycin], a1-acid glycoprotein (AAG), and dextran were purchased from Sigma–Aldrich Corporation (St. Louis, MO). Human serum albumin (HSA), 20%, was obtained from Dong-Shin Pharmaceutical Company (Seoul, South Korea). Various buffer solutions having pHs 1 and 2 (HCl-KCl buffer), pH 3 (KHC8H4O4-HCl buffer), pHs 4 and 5 (KHC8H4O8-NaOH buffer), pHs 6 and 7 (KH2PO4-NaOH buffer), pHs 8 and 9 (H3BO3-KCl-NaOH buffer), pH 10 (NaHCO3-NaOH buffer), pH 11 (Na2HPO4-NaOH buffer), and pHs 12 and 13 (KCl-NaOH buffer) were products from Shinyo Pure Chemicals (Osaka, Japan). Other chemicals were of reagent grade or HPLC grade.
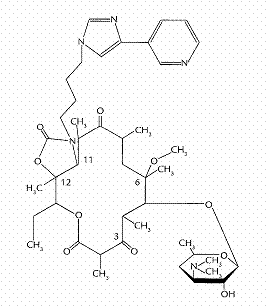
Figure 1: Chemical structure of telithromycin.
Animals
Male Sprague–Dawley rats of 6–7 weeks of age (weighing 200–250 g) were purchased from Taconic Farms Inc. (Samtako Bio Korea, O-San, South Korea). All rats were maintained in a clean room (Animal Center for Pharmaceutical Research, College of Pharmacy, Seoul National University, Seoul, South Korea) at a temperature of between 20 and 23 °C with 12-h light (0700–1900) and dark (1900–0700) cycles and a relative humidity of 50 ± 5%. Rats were housed in metabolic cages (Tecniplast, Varese, Italy) under the supply of filtered pathogen-free air and with food (Agribrands Purina Korea, Pyeongtaek, South Korea) and water ad libitum. Animal Care and Use Committee of College of Pharmacy of Seoul National University approved the protocol of this animal study.
Intravenous and Oral Administration of Telithromycin to Rats
The pretreatment and surgical procedures for intravenous and oral administration were similar to the previously reported methods (8). In the early morning, the jugular vein (for drug administration only for intravenous study) and the carotid artery (for blood sampling) of each rat were cannulated with a polyethylene tube (Clay Adams, Parsippany, NJ) under light ether anesthesia. Both cannulas were exteriorized to the dorsal side of the neck, where each cannula was terminated with a long silastic tube (Dow Corning, Midland, MI). Both silastic tubes were covered with a wire sheath to allow free movement of the rats. After the exposed areas were surgically sutured, each rat was housed individually in a rat metabolic cage (Daejong Scientific Company, Seoul, South Korea) and allowed to recover from the anesthesia for 4 to 5 h before the study began. They were not restrained during the whole experimental period. Telithromycin (dissolved in distilled water adjusted to pH 3 with acetic acid) at doses of 20 (n = 9), 50 (n = 8), and 100 (n = 6) mg/kg was administered via the jugular vein over 1 min (total infusion volume of approximately 0.6 mL) to rats. An approximately 220 mL aliquot of blood sample was collected via the carotid artery at 0 (to serve as a control), 1 (at the end of the infusion), 5, 15, 30, 60, 120, 180, 240, 360, 480, 600, and 720 min after the start of the intravenous infusion of telithromycin. After centrifugation of blood sample, a 100 mL aliquot of plasma sample was stored in a -70 ºC freezer (Model DF8517; Ilshin Laboratory Company, Seoul, South Korea) until the HPLC analysis of telithromycin (9). An approximately 0.3 mL aliquot of the heparinized 0.9% NaCl-injectable solution (20 m/mL) was used to flush each cannula immediately after each blood sampling to prevent blood clotting. At the end of the experiment (24 h), each metabolic cage was rinsed with 10 mL of distilled water and the rinsings were combined with 24-h urine. After measuring the exact volume of the combined urine sample, two 100 mL aliquots of the combined urine sample were stored in a –70 ºC freezer until HPLC analysis of telithromycin (9). At the same time (24 h), each rat was exsanguinated and sacrificed through cervical dislocation. The entire gastrointestinal tract (including its contents and feces) of each rat was removed, transferred into a beaker that contained 100 mL of methanol (to facilitate the extraction of telithromycin), and cut into small pieces using scissors. After manual shaking and stirring with a glass rod for 1 min, two 100 mL aliquots of the supernatant were collected from each beaker and stored in a –70 ºC freezer until HPLC analysis of telithromycin (9).
Telithromycin (the same solution that was used in the intravenous study) at doses of 20 (n = 8), 50 (n = 6), and 100 (n = 7) mg/kg was administered orally (total oral volume of approximately 1.5 mL) in rats using a feeding tube after overnight fasting with free access to water. Blood samples were collected via the carotid artery at 0, 5, 15, 30, 45, 60, 90, 120, 180, 240, 360, 480, 600, and 720 min after the oral administration of telithromycin. Other procedures including the plasma, urine, and gastrointestinal samples were similar to those in the intravenous study.
Hepatic First-Pass Effect of Telithromycin in Rats
The carotid artery and the jugular vein of each rat were cannulated under light ether anesthesia (8). At the same time, the vein from the ceacum was cannulated and the cannula was pushed forward about 4.0 cm toward the liver through the portal vein to minimize impaired blood flowing into the portal vein (10,11). Telithromycin (the same solution that was used in the intravenous study was diluted in distilled water and the concentrations were adjusted to rats’ body weight) at a dose of 50 mg/kg was infused (approximately 2 mL/kg) over 30 min at a rate of 2 mL/h into the jugular vein and the portal vein for intravenous (n = 5) and intraportal (n = 5) administration, respectively, with the assistance of an infusion pump (model 2400-006; Harvard Instrument, Southnatick, MA). At the same time, an equal volume of the distilled water adjusted to pH 3 with acetic acid was also infused over 30 min via the portal vein for intravenous study and via the jugular vein for intraportal study. Blood samples were collected via the carotid artery at 0, 15, 30 (at the end of the infusion), 31, 35, 45, 90, 150, 270, 390, 510, 630, and 750 min after the start of the intravenous infusion of telithromycin.
Gastric and Intestinal First-Pass Effects of Telithromycin in Rats
Rats were fasted overnight with free access to water. The carotid artery and the vein from the ceacum of each rat were cannulated (10,11). For intraportal infusion (n = 5), telithromycin (the same solution that was used in the intravenous study) at a dose of 50 mg/kg was infused (approximately 0.6 mL) via the portal vein over 30 min, and an equal volume of the distilled water adjusted to pH 3 with acetic acid was instilled into the stomach and duodenum, respectively, using a 23 gauge needle. For intraduodenal instillation (n = 4), an equal volume of the distilled water adjusted to pH 3 with acetic acid was instilled into the stomach and infused via the portal vein over 30 min, respectively, and telithromycin at a dose of 50 mg/kg was instilled into the duodenum. For intragastric instillation (n = 5), an equal volume of the distilled water adjusted to pH 3 with acetic acid was instilled into the duodenum and infused via the portal vein over 30 min, respectively, and telithromycin at a dose of 50 mg/kg was instilled into the stomach. Blood samples were collected via the carotid artery at 0, 15, 30, 31, 35, 45, 90, 150, 270, 390, 510, 630, and 750 min after the start of the intraportal infusion of telithromycin, and 0, 15, 30, 60, 120, 180, 240, 360, 480, 600, 720, 840, 960, and 1,080 min after the intragastric and intraduodenal instillation of telithromycin.
Stability of Telithromycin
The procedures for the stability of telithromycin were similar to the previously reported methods (12). Telithromycin stock solution (dissolved in distilled water adjusted to pH 3 with acetic acid) was spiked (10 mL per mL) in each test tube that contained three rat gastric juices (pHs of 1, 2.5, and 3, respectively) and various buffer solutions having pHs ranging from 1 to 13 to produce a telithromycin concentration of 0.5 mg/mL. Various buffer solutions were incubated in a water-bath shaker kept at temperature of 37 ± 2 °C and at a rate of 50 oscillations per min (opm) for 48 h and three rat gastric juices for 4 h. The concentrations of telithromycin in the above samples were analyzed using the reported HPLC method (9) as soon as the samples were collected.
In vitro Distribution Kinetics of Telithromycin between Plasma and Blood Cells of Rat Blood
The procedures for the in vitro distribution kinetics of telithromycin between plasma and blood cells of rat blood were similar to the previously reported methods (13). One milliliter of heparinized blood (freshly withdrawn via the carotid artery from seven unanesthetized rats; the rat blood was pooled together) was pipetted into each glass test tube (22 tubes for each concentration). The telithromycin stock solutions (the same solution that was used in the intravenous study) having the concentrations of 0.1, 0.5, and 1 mg/mL, respectively, were spiked (10 µL) into above each glass test tube to make the final concentrations of 1, 5, and 10 mg/mL, respectively. At 0, 1, 3, 5, 7, 10, 15, 30, 60, 90, and 120 min, blood samples were centrifuged and plasma samples were collected. Whole blood concentrations of telithromycin were also measured by adding 2 volumes of distilled water to facilitate the hemolysis and to increase the reproducibility of HPLC assay of telithromycin (13).
Factors Influencing the Binding of Telithromycin to 4% HSA Using the Equilibrium Dialysis Technique
The procedures for the protein binding of telithromycin to 4% HSA were similar to the previously reported methods (14). One milliliter of 4% HSA (20% HSA was diluted with isotonic Sørensen phosphate buffer of pH 7.4) was dialyzed against 1 mL of an isotonic Sørensen phosphate buffer of pH 7.4 that contained 3% (w/v) dextran (‘the buffer’) in a 1 mL dialysis cell (Spectrum Medical Industries, Los Angeles, CA) using a Spectra/Por 4 membrane (molecular weight cutoff of 12,000–14,000 Dalton; Spectrum Medical Industries). Each cell was incubated in a water-bath shaker kept at 37 °C and at a rate of 50 opm. After 24 h incubation, two 0.1 mL aliquots were collected from each compartment and stored in a –70 °C freezer until HPLC analysis of telithromycin (9). The effects of telithromycin and HSA concentrations, incubation temperature, various buffers, buffer pHs, amounts of heparin and AAG, and other drugs which are used widely in clinic were also evaluated (14). The binding of telithromycin to fresh rat plasma (n = 3) was also measured at a telithromycin concentration of 5 mg/mL.
HPLC Analysis of Telithromycin
The concentrations of telithromycin in the above samples were determined by a slight modification of the reported HPLC method (9); quinine hydrochloride instead of RU 66260 was used as an internal standard. To a 100 mL aliquot of biological sample, a 25 mL aliquot of distilled water that contained a 100 mg/mL of quinine hydrochloride (an internal standard) and a 300 mL aliquot of acetonitrile were added. After vortex-centrifugation at 12000 rpm for 5 min, the organic layer was collected and dried (Dry thermobath, Eyela, Tokyo, Japan) under a gentle stream of nitrogen gas at 40 oC. A 100 mL aliquot of the mobile phase was added to reconstitute the residue and a 50 mL aliquot was injected directly onto a reversed-phase HPLC column. The mobile phase, ammonium acetate (0.05 M) : methanol : acetonitrile (49.5:27.6:22.9, v/v/v), was run at a flow-rate of 0.9 mL/min, and the column effluent was monitored using a fluorescence detector. The retention times of the internal standard (quinine hydrochloride) and telithromycin were approximately 4.3 and 9.5 min, respectively. The detection limits of telithromycin in rat plasma, urine, and gastrointestinal samples were all 0.1 mg/mL. The coefficients of variation of the assay were below 5.70, 7.94, and 6.31% for rat plasma, urine, and gastrointestinal samples, respectively.
The HPLC system consisted of a model 717 autosampler (Waters Corporation, Milford, MA), a model L-6000 pump (Hitachi, Tokyo, Japan), reversed-phase column (C18; 15 cm, l. ´ 4.6 mm, i.d.; particle size, 5 µm; CAPCELL PAK, Shiseido, Tokyo, Japan), fluorescence detector (SpectraSYSTEM® FL3000, Thermo Scientific, MA) set at an excitation wavelength of 264 nm and an emission wavelength of 460 nm at room temperature, and data system (Autochro-2000, Young Lin Instrument Co., Ltd., Seoul, South Korea).
Pharmacokinetic Analysis
The AUC was calculated using the trapezoidal rule–extrapolation method; this method uses the logarithmic trapezoidal rule for the calculation of the area during the declining plasma–level phase (15) and the linear trapezoidal rule for the rising plasma–level phase. The area from the last datum point to time infinity was estimated by dividing the last measured plasma concentration by the terminal-phase rate constant.
Standard methods (16) were used to calculate the following pharmacokinetic parameters using the noncompartmental analysis (WinNonlin®, Pharsight Corporation, Mountain View, CA); the time-averaged total body clearance (CL), renal clearance (CLR), and nonrenal clearance (CLNR), terminal half-life, first moment of AUC (AUMC), mean residence time (MRT), apparent volume of distribution at steady state (Vdss), and F (8). The peak plasma concentration (Cmax) and time to reach a Cmax (Tmax) were directly read from the experimental data.
The mean values of each clearance (17), terminal half-life (18), and Vdss (19) were calculated using the harmonic mean method.
Statistical Analysis
A P-value of less than 0.05 was considered to be statistically significant using a t-test between the two means for the unpaired data, or a Duncan’s multiple range test of Statistical Package for the Social Sciences (SPSS) posteriori analysis of variance (ANOVA) among the three means for the unpaired data. All results are expressed as means ± standard deviation except medians (ranges) for Tmax.
RESULTS
Intravenous and Oral Administration of Telithromycin to Rats
After intravenous infusion of telithromycin at doses of 20, 50, and 100 mg/kg to rats, the mean arterial plasma concentration–time profiles of the drug are shown in Figure 2(A), and some relevant pharmacokinetic parameters are listed in Table 1. The mean arterial plasma concentrations of telithromycin declined in a polyexponential manner for all three intravenous doses studied. Note that the dose-normalized (based on 10 mg/kg) AUC values of telithromycin were dependent on three intravenous doses of the drug; the values were 241 ± 22.8, 440 ± 52.9, and 592 ± 52.8 µg min/mL for 20, 50, and 100 mg/kg, respectively; each value was significantly different. Moreover, the slope between log AUC values and log doses of telithromycin was greater than 1.00 (the value was 1.56). Similar results were also obtained from CL and CLNR; the CL and CLNR were significantly slower with increasing intravenous doses. At 100 mg/kg, the terminal half-life and MRT were significantly longer and Vdsswas significantly larger than those at 20 and 50 mg/kg (Table 1). The CLR at 20 mg/kg was significantly faster than those at 50 and 100 mg/kg (106 and 144% increase, respectively). However, the percentages of intravenous dose of telithromycin excreted in 24 h urine as an unchanged drug (Ae0–24 h) and recovered from the entire gastrointestinal tract (including its contents and feces) at 24 h as an unchanged drug (GI24 h) were not significantly different among three intravenous doses studied. The above data indicate that the pharmacokinetic parameters of telithromycin depend on three intravenous doses studied in rats.
After oral administration of telithromycin at doses of 20, 50, and 100 mg/kg to rats, the mean arterial plasma concentration–time profiles of the drug are shown in Figure 2(B), and some relevant pharmacokinetic parameters are also listed in Table 1. After oral administration of telithromycin, absorption of the drug from the rat gastrointestinal tract was rapid; telithromycin was detected in plasma from the first blood sampling time (5 min) for the three oral doses studied. After reaching Tmax, the plasma concentrations of telithromycin declined in a monoexponential manner for three oral doses studied. Note that the dose-normalized (based on 10 mg/kg) AUC values of telithromycin were also dependent on three oral doses of the drug; the values were 65.6 ± 24.4, 121 ± 46.0, and 227 ± 39.3 mg min/mL for 20, 50, and 100 mg/kg, respectively; each value was significantly different (Table 1). Moreover, the slope between log AUC values and log doses of telithromycin was greater than 1.00 (the value was 1.77). The terminal half-life at 100 mg/kg was significantly longer than those at 20 and 50 mg/kg (75.7 and 75.7% increase, respectively), and the dose-normalized (based on 10 mg/kg) Cmax at 20 mg/kg was significantly lower than those at 50 and 100 mg/kg (57.6 and 55.8% decrease, respectively) (Table 1). The Ae0–24 h at 20 mg/kg was significantly smaller (54.8% decrease) than that at 50 mg/kg (Table 1). However, the Tmax, CLR, and GI24 h were not significantly different among three oral doses studied (Table 1). The F values were 27.1, 27.5, and 38.4% for oral doses of 20, 50, and 100 mg/kg, respectively (Table 1). The above data indicate that the pharmacokinetic parameters of telithromycin also depend on three oral doses studied in rats.
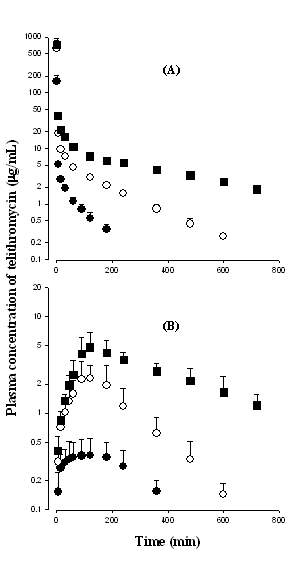
Figure 2: Mean arterial plasma concentration–time profiles of telithromycin after 1 min intravenous infusion of the drug at doses of 20 (●; n = 9), 50 (○; n = 8) and 100 (■; n = 6) mg/kg (A), and oral administration of the drug at doses of 20 (●; n = 8), 50 (○; n = 6) and 100 (■; n = 7) mg/kg (B) to rats. Bars represent standard deviation.
Hepatic First-Pass Effect of Telithromycin in Rats
After intravenous and intraportal administration of telithromycin, the mean arterial plasma concentration–time profiles of the drug are shown in Figure 3(A). The AUC values of telithromycin at a dose of 50 mg/kg were not significantly different between intravenous and intraportal administration (2980 ± 776 and 2930 ± 1320 mg min/mL), suggesting that the hepatic first-pass effect of telithromycin after absorption into the portal vein is almost negligible, if any, in rats.
Gastric and Intestinal First-Pass Effects of Telithromycin in Rats
After intraportal, intragastric, and intraduodenal administration of telithromycin, the mean arterial plasma concentration–time profiles of the drug are shown in Figure 3(B). The AUC values of telithromycin at a dose of 50 mg/kg were not significantly different between intragastric and intraduodenal administration (898 ± 161 and 1010 ± 486 mg min/mL), however, the values were significantly smaller than that after intraportal administration (2980 ± 1090 mg min/mL). This suggests that the gastric first-pass effect of telithromycin is almost negligible, if any, and the intestinal first-pass effect of telithromycin was approximately 66.1% in rats.
Stability of Telithromycin
Stabilities of telithromycin in three rat gastric juices and various buffer solutions are listed in Table 2. Telithromycin was stable in three rat gastric juices (more than 90.5% of the spiked amounts of the drug was recovered after 4 h incubation in three rat gastric juices), and various buffer solutions having pHs ranging from 1 to 13 (more than 92.0% was recovered) up to 48 h incubation except at pH 4 (83.7% was recovered after 48 h incubation). The above data suggest that telithromycin is relatively stable against chemical (except at pH 4) and enzymatic degradation.
In vitro Distribution Kinetics of Telithromycin between Plasma and Blood Cells of Rat Blood
Telithromycin was stable when incubated in rat whole blood; the whole blood and plasma concentrations of telithromycin were almost constant up to 2 h incubation (data not shown). This indicates that telithromycin reached equilibrium rapidly (within 30 s mixing manually) between plasma and blood cells of rat blood. The equilibrium plasma-to-blood cells partition ratios of telithromycin were independent of initial blood telithromycin concentrations of 1, 5, and 10 mg/mL; the mean value was 0.588 (range 0.404–0.965).
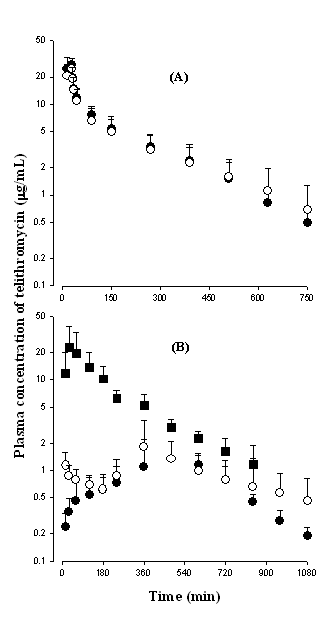
Figure 3: Mean arterial plasma concentration–time profiles of telithromycin after intravenous (●; n = 5) and intraportal (○; n = 5) administration of the drug (A), and intragastric (●; n = 5), intraduodenal (○; n = 4), and intraportal (■; n = 5) administrations of the drug (B) at a dose of 50 mg/kg to rats. Bars represent standard deviation.
Factors Influencing the Binding of Telithromycin to 4% HSA Using the Equilibrium Dialysis Technique
Equilibrium of telithromycin between 4% HSA and ‘the buffer’ compartments was reached after 12 h incubation and the binding values were not affected up to 24 h incubation; the mean binding value was 26.4 ± 1.51% at telithromycin concentrations ranging from 1 to 10 mg/mL. However, the values were 20.5, 18.1, and 17.9% at telithromycin concentrations of 20, 50, and 100 mg/mL, respectively. This could be due to the limited binding sites in HSA. Hence, in the subsequent protein binding studies, 24-h incubation and a telithromycin concentration of 5 mg/mL were employed. The binding of telithromycin seemed to be dependent on the concentrations of salicylic acid (37.4 and 40.0% for 150 and 300 mg/mL, respectively) and AAG (68.7, 78.8, and 84.4% for 0.08, 0.16, and 0.32%, respectively), incubation temperatures (34.4, 28.5, and 27.1% for 4, 22, and 37 °C, respectively), and various buffers (27.1, 31.7, 34.2, and 27.8% for isotonic Sørensen phosphate buffer of pH 7.4, distilled water, 0.9% NaCl-injectable solution, and 5% glucose, respectively). However, the buffer pHs (pHs of 5.8, 7.0, 7.4, and 8.0), and concentrations of HSA (0.5, 1, 2, 3, and 4%), heparin (10 and 40 units/mL), and sulfisoxazole (100, 200, and 300 mg/mL) did not influence the binding of telithromycin. The binding value of telithromycin to fresh rat plasma (n = 3) was 71.6 ± 5.31% at a telithromycin concentration of 5 mg/mL. This value, 71.6%, was greater than 26.4% to 4% HSA. This could be due to greater binding of telithromycin, a basic drug, to AAG.
discussion
In pharmacokinetic studies, accurately measured plasma drug concentrations are usually assumed to be equal to their in vivo plasma concentrations. Such an assumption may be valid for drugs that have very rapid or instantaneous rates of equilibration between plasma and blood cells (13,20). If this equilibration process is slow or irregular, then the length of time elapsed between collection and centrifugation of a blood sample might have a profound effect on the measured drug concentration (“the blood storage effect”); the sooner the centrifugation, the higher will be the plasma concentration measured (13,20). This factor might have contributed in part to the ‘reported’ inconsistencies in the time to achieve the peak plasma level after intravenous administration, to the ‘calculated’ time-dependent renal clearances, and to the ‘unsmooth’ plasma level profiles reported in the literature (20). Moreover, it was reported that the bound fractions of adriamycin (21) and propranolol (22) to red blood cells act as barriers for their elimination. Hence, the experiments on the distribution kinetics of telithromycin between plasma and blood cells of rat blood were performed.
It was reported that the binding of drugs to plasma proteins was dependent on the incubation temperature (the binding force decreases with increasing temperature), chloride ion in the buffer solutions (quinidine and azosemide bind to chloride), buffer pHs (unionized forms of drugs have higher affinity to proteins than that of ionized forms of drugs), concentrations of proteins (due to limited binding sites in proteins), heparin (heparin influences the protein binding of imipramine), AAG (especially for basic drugs), and other drugs frequently used in medical practice (sulfisoxazole and salicylic acid) (14). Hence, these factors were evaluated in the present study.
After intravenous administration of telithromycin, the contribution of CLR to CL of the drug was not considerable; the Ae0–24 h values were less than 10.5% of intravenous dose of the drug for all three doses studied (Table 1), suggesting that most of the intravenously administered telithromycin are eliminated via the nonrenal route (CLNR). The contribution of gastrointestinal (including biliary) excretion of an unchanged telithromycin to CLNR of the drug also was not considerable; the GI24 h values were less than 7.11% of intravenous dose of the drug for all three doses studied (Table 1). The smaller GI24 h values, less than 7.11% (Table 1), were not likely due to chemical and enzymatic degradation of telithromycin in rats’ gastric fluids; telithromycin was relatively stable in various buffer solutions and rat gastric juices as mentioned earlier. For the exact measurement of the GI24 h (including its contents and feces), the stability test in rat gastric juices and various buffer solutions is required. The above data suggest that the CLNR of telithromycin listed in Table 1 could represent the metabolic clearance of the drug. Hence, the changes in CLNR of telithromycin could be due to the changes in the metabolic clearance of the drug in rats.
After both intravenous and oral administration of telithromycin, the dose-normalized AUC values increased with increasing doses; this could be mainly due to saturation of metabolism of telithromycin in rats and this could be supported by significantly slower CLNR of telithromycin with increasing doses (Table 1). Recently, it was reported from our laboratories that telithromycin is mainly metabolized via the hepatic microsomal cytochrome P450 (CYP) 3A1/2 in male Sprague–Dawley rats (23). For example, in rats pretreated with dexamethasone phosphate (a main inducer of CYP3A1/2 in rats), the CLNR of telithromycin was significantly faster (129% increase) than the controls. And in rats pretreated with troleandomycin (a main inhibitor of CYP3A1/2 in rats), the CLNR of telithromycin was significantly slower (31.3% decrease) than the controls. The dose-dependent AUC of telithromycin after oral administration of the drug was also reported in humans; the dose-normalized AUC values increased with increasing single oral doses, 400, 800, and 1600 mg (7).
The renal extraction ratios of telithromycin (CLR of telithromycin / kidney plasma flow rate; only for urinary excretion of unchanged drug) were estimated based on the CLR of telithromycin (Table 1), reported kidney blood flow rate of 36.8 mL/min/kg (24), and hematocrit of approximately 45% (25) in rats. The values thus estimated were 19.6, 9.49, and 8.00% for 20, 50, and 100 mg/kg, respectively. The above data indicate that telithromycin is excreted poorly via rat kidney (a low renal extraction ratio drug).
Although the F values of telithromycin were different between humans, 57% (4–6) and rats, 27.1–38.4% (Table 1), measuring the first-pass effects of the drug in humans is not quite easy. Hence, rats were used as an animal model in this study.
The F of telithromycin at an oral dose of 50 mg/kg was 27.5% (Table 1). After oral administration of telithromycin at a dose of 50 mg/kg, the GI24 h was 5.85% of oral dose (Table 1). It is possible that this unchanged telithromycin, 5.85%, might be partly attributed to the gastrointestinal excretion (including biliary excretion) of the absorbed drug. For comparison, the mean “true” fraction of dose unabsorbed (Funabs) at an oral dose of 50 mg/kg could be estimated by the previously reported equation (26);
0.0585 = Funabs + (0.275 ´ 0.0651) (1)
in which 0.275 and 0.0651 are the F and GI24 h after intravenous administration of telithromycin at a dose of 50 mg/kg, respectively (Table 1). The Funabs thus calculated was 4.06%, indicating that the contribution of gastrointestinal (including biliary) excretion of the absorbed telithromycin to the total drug recovered from the gastrointestinal tract following oral administration of the drug was almost negligible, 1.79%. Hence, approximately 96% of oral dose of telithromycin at 50 mg/kg was absorbed from rat gastrointestinal tract. Since 4.06% of orally administered telithromycin at a dose of 50 mg/kg was not absorbed from rat gastrointestinal tract up to 24 h and F value was 27.5%, approximately 68.4% (100% – 4.06% – 27.5%) of orally administered telithromycin at a dose of 50 mg/kg could be eliminated by the first-pass effect in rats.
After intravenous administration of telithromycin, the CL, 16.9–41.5 mL/min/kg based on plasma data (Table 1), were considerably slower than the reported cardiac output of 296 mL/min/kg based on blood data (24) and hematocrit of approximately 45% (25) in rats. The above data suggest that the first-pass effect of telithromycin in the lung and heart could be almost negligible, if any, in rats.
After intragastric and intraduodunal instillation of telithromycin at a dose of 50 mg/kg to rats, the AUC values of the drug were not significantly different between two routes of administration, suggesting that gastric first-pass effect of telithromycin is almost negligible, if any, in rats. However, the AUC after intraduodenal administration of telithromycin at a dose of 50 mg/kg was 33.9% of that after intraportal administration, suggesting that intestinal first-pass effect of telithromycin was approximately 66.1%. The approximately 66.1% is equivalent to 63.4% of oral dose, considering the unabsorbed fraction, 4.06%. Telithromycin is reported to be metabolized to RU 76363, an alcohol resulting from loss of aryl rings (major hepatic metabolite of telithromycin) (7). It was reported that CYP3A is mostly expressed in rat intestine (27). Although what kinds of CYP systems are involved for the metabolism of telithromycin in rat intestine is not known, the studies on the effect of intestinal CYP3A1/2 for the metabolism of telithromycin is required.
Therefore, it could be concluded that approximately 32.5% (100% – 63.4% – 4.06%) of orally administered telithromycin at a dose of 50 mg/kg could be absorbed into the portal vein. After intraportal administration of telithromycin at a dose of 50 mg/kg to rats, the AUC of telithromycin was comparable to that after intravenous administration at a dose of 50 mg/kg. Hence, the hepatic first-pass effect of telithromycin after absorption into the portal vein could be almost negligible, if any, in rats. Hence, the value of 32.5% is close to the F value of 27.5% (Table 1).
The considerable intestinal first-pass effects were also reported from other drugs. For example, furosemide, azosemide, YH439 (a new hepatoprotective agent), YJA-20379-8 (a new reversible proton pump inhibitor), iprifavone, bumetanide, KR-31543 (a new neuroprotective agent for ischemia-reperfusion damage), SR-4668 (a candidate for diabetic neuropathy), KR-60436 (a new reversible proton pump inhibitor), and oltipraz in rats, and midazolam and saquinavir in humans have been reported (12 and references therein).
CONCLUSIONS
After oral administration of telithromycin at a dose of 50 mg/kg, the unabsorbed fraction was approximately 4.06% of the dose, F was 27.5%, and intestinal first-pass effect was approximately 63.4% of the dose in rats. The low F of telithromycin at an oral dose of 50 mg/kg was mainly due to considerable intestinal first-pass effect (approximately 63.4% of an oral dose) in rats. Since the dose-normalized AUC values of oral telithromycin increased with increasing doses (Table 1), the intestinal first-pass effects of telithromycin are expected to be changed with different oral doses in rats. However, the changes seemed not to be considerable, because the F values were low; 27.1, 27.5, and 38.4% for doses of 20, 50, and 100 mg/kg, respectively (Table 1).
ACKNOWLEDGMENTS
The authors thank to Dr. Jürgan Pünter of Aventis Pharma Deutschland GmbH (Frankfurt am Main, Germany) for the kind donation of telithromycin. This study was supported in part by a grant from the Seoul City Collaborative Project among the Industry, Academy, and Research Institute, Korea.
Table 1: Mean (± standard deviation) pharmacokinetic parameters of telithromycin after 1 min intravenous infusion and oral administration of the drug at various doses to rats.
Parameter |
Intravenous |
|
Oral |
||||||||||||||||
20 mg/kg (n = 9) |
50 mg/kg (n = 8) |
100 mg/kg (n = 6) |
|
20 mg/kg (n =8) |
50 mg/kg (n =6) |
100 mg/kg (n =7) |
|||||||||||||
Body weight (g) |
258 |
± |
18.0a |
278 |
± |
7.07 |
238 |
± |
10.3 |
|
236 |
± |
12.5 |
237 |
± |
4.08 |
239 |
± |
2.44 |
AUCb (mg min/mL) |
482 |
± |
45.7a |
2200 |
± |
265 |
5920 |
± |
528 |
|
131 |
± |
48.7a |
605 |
± |
230 |
2270 |
± |
393 |
Terminal half-life (min) |
67.1 |
± |
18.4 |
125 |
± |
22.8 |
257 |
± |
119 |
|
115 |
± |
58.5 |
115 |
± |
62.3 |
202 |
± |
127d |
MRT (min) |
43.7 |
± |
16.2 |
92.5 |
± |
17.1 |
311 |
± |
94.5 |
|
|
|
|
|
|
|
|
|
|
Cmaxb (mg/mL) |
|
|
|
|
|
|
|
|
|
|
0.488 |
± |
0.171e |
2.88 |
± |
0.986 |
5.51 |
± |
1.54 |
Tmaxd (min) |
|
|
|
|
|
|
|
|
|
|
90 (15–120) |
105 (90–180) |
120 (90–360) |
||||||
CL (mL/min/kg) |
41.5 |
± |
3.93a |
22.8 |
± |
2.76 |
16.9 |
± |
1.68 |
|
|
|
|
|
|
|
|
|
|
CLR (mL/min/kg) |
3.96 |
± |
1.05e |
1.92 |
± |
0.904 |
1.62 |
± |
0.840 |
|
5.21 |
± |
5.28 |
6.87 |
± |
2.69 |
2.72 |
± |
3.96 |
CLNR (mL/min/kg) |
38.0 |
± |
3.62a |
20.5 |
± |
2.94 |
15.1 |
± |
1.16 |
|
|
|
|
|
|
|
|
|
|
Vdss (mL/kg) |
1630 |
± |
815 |
2010 |
± |
605 |
4980 |
± |
1230 |
|
|
|
|
|
|
|
|
|
|
Ae0–24 h (% of dose) |
9.86 |
± |
2.49 |
9.99 |
± |
3.55 |
10.5 |
± |
3.36 |
|
3.83 |
± |
0.857f |
8.48 |
± |
3.25 |
6.60 |
± |
2.60 |
GI24 h (% of dose) |
4.53 |
± |
1.58 |
6.51 |
± |
3.89 |
7.11 |
± |
3.04 |
|
11.1 |
± |
7.04 |
5.85 |
± |
3.94 |
10.7 |
± |
5.95 |
F (%) |
|
|
|
|
|
|
|
|
27.1 |
27.5 |
38.4 |
||||||||
a Each value was significantly different (p < 0.05).
b AUC and Cmax values were normalized to the telithromycin dose of 10 mg/kg for statistical analysis.
c 100 mg/kg was significantly different (p < 0.05) from 20 and 50 mg/kg.
dTmax was expressed as median (ranges).
e 20 mg/kg was significantly different (p < 0.05) from 50 and 100 mg/kg.
f 20 mg/kg was significantly different (p < 0.05) from 50 mg/kg.
Table 2: Stability of telithromycin
|
Rat gastric juice up to 4 h incubation |
|||||||||||||
|
Rat 1 (pH = 1.0) |
Rat 2 (pH = 2.5) |
Rat 3 (pH = 3.0) |
|||||||||||
Recovery (%) |
90.5 |
96.0 |
91.9 |
|||||||||||
|
Buffer pHs up to 48 h incubation |
|||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
|
Recovery (%) |
97.4 |
103 |
95.0 |
83.7 |
99.7 |
99.6 |
105 |
93.1 |
92.0 |
100 |
97.3 |
102 |
110 |
|