J Pharm Pharmaceut Sci (www.cspscanada.org) 10(1):71-85, 2007
Difference between Pharma- cokinetics of Mycophenolic acid (MPA) in Rats and that in Humans is caused by Different affinities of MRP2 to a glucuronized form
Yoh Takekuma1, Haruka Kakiuchi2, Koujiro Yamazaki2, Seiji Miyauchi3, Takashi Kikukawa4, Naoki Kamo3, Vadivel Ganapathy5, Mitsuru Sugawara2
1 Laboratory of Pharmcotherapeutic Information, Faculty of Pharmaceutical Science, Hokkaido University, Sapporo, Japan 2 Department of Pharmacy, Hokkaido University Hospital, Sapporo, Japan 3 Laboratory of Biophysical Chemistry, Faculty of Pharmaceutical Science, Hokkaido University, Sapporo, Japan 4 Laboratory of Biomolecular Systems, Creative Research Initiative “Sosei” (CRIS), Hokkaido University, Sapporo, Japan 5 Department of Biochemistry and Molecular Biology, Medical College of Georgia, Augusta, Georgia, USA
Received December 17, 2007, Revised February 20, 2007, Accepted April 20, 2007, Published, April 22, 2007
Corresponding Author: Dr. Yoh Takekuma, Faculty of Pharmaceutical Science, Hokkaido University, Kita-12-jo, Nishi-6-chome, Kita-ku, Sapporo 060-0812, Japan. E-mail: y-kuma@pharm.hokudai.ac.jp
ABSTRACT - PURPOSE. Mycophenolic acid (MPA), an immunosuppressant, is excreted as its glucuronized form, MPAG. In humans, MPAG is mostly excreted into urine, whereas more than 80% of the dose is excreted into bile in rats. The aim of this study was to clarify the cause of the species difference. We investigated whether MPAG is a substrate of human organic anion transporters (hOATs), and we compared the affinities of multi-drug resistance-associated protein 2 (MRP2) for MPAG in rats and humans. METHODS. The inhibitory effects of MPAG on the uptake of typical substrates via hOAT1 and hOAT3 were determined using HeLa cells heterologously expressing hOAT1 and Xenopus laevis oocytes heterologously expressing hOAT3. MPAG transport activity via hOAT1 and hOAT3 was determined by the two-microelectrode voltage-clamp technique using Xenopus laevis oocytes expressing hOAT1 and hOAT3. The affinities of MPAG for hMRP2 and rMrp2 were determined by the inhibitory effects of MPAG on p-aminohippuric acid (a typical substrate) uptake using membrane vesicles expressing hMRP2 or rMrp2. RESULTS. MPAG inhibited the uptake of PAH via hOAT1 and hOAT3, and calculated IC50 values were 222.6±26.6 µM and 41.5±11.5 µM, respectively. However, MPAG was not transported by hOAT1 and hOAT3. MPAG strongly inhibited the uptake of PAH via both rMrp2 and hMRP2. However, the magnitudes of inhibitory effects were different. The calculated IC50 values were 286.2±157.3 µM and 1036.8±330.5 µM, respectively. CONCLUSION. MPAG is not a substrate but is an inhibitor of hOAT1 and hOAT3. The affinity of rMRP2 to MPAG was about 3.6 times as high as that of hMRP2. Therefore, the difference of affinity between hMRP2 and rMrp2 is a possible mechanism of the difference of excretion ratio of MPAG between rats and human.
introduction
Mycophenolate mofetil (MMF) is an ester prodrug of mycophenolic acid (MPA), an immunosuppressant. It has been shown that MPA selectively inhibits inosine monophosphate dehydrogenase, and MMF has been widely used for the prevention of rejection in patients receiving allogeneic transplants [1]. The use of MMF has improved therapeutic outcome for transplantations [2-5]. However, interindividual differences of blood concentrations in MPA are very large [6-9]. Therefore, it is important to clarify the cause of this difference for individualized therapy.
Orally administered MMF is hydrolyzed to MPA through the process of absorption from the gastrointestinal tract. MPA is metabolized to two kinds of glucuronidated metabolites. One is a glucuronide of the phenolic hydroxyl group (MPAG) and the other is a glucuronide of the acylic hydroxyl group. The former is a major metabolite and the latter is a minor metabolite [6]. MPAG has no immunosuppressive activity. However, because MPAG is excreted into bile and re-absorbed as MPA by enterohepatic circulation, its variation affects the magnitude of immunosuppressive activity [6]. There are differences between pharmacokinetics of MPA in rats and humans. Although the main metabolite (MPAG) is the same in humans and rats, it has known that pharmacokinetics of MPA between humans and rats were different greatly. In humans, MPAG is mostly excreted into urine (about 71% of the MMF dose in 48 hour after oral administration), and area under the concentration-time curve (AUC) of MPA after oral administration of MMF is smaller than that of MPAG [10]. In contrast, in rats, 84% of the dose is excreted into bile, and AUC of MPA is larger than that of MPAG [11, 12]. It has been reported that MPAG is excreted into bile by multi-drug resistance-associated protein 2 (MRP2) [13]. Therefore, it is possible that this species variability were caused by different affinities of MRP2.
On the other hand, it is also possible that MPAG is excreted into urine by an active transporter in humans. Human organic anion transporters (hOATs) play an important role in the urinary excretion and reabsorption of drugs [14-16]. Since MPAG is an organic anion, it is possible that it is a substrate of hOATs. Furthermore, it is thought that clarifying these differences is useful to analyze the factor of variability of pharmacokinetics in humans.
In this study, we investigated whether MPAG is a substrate of hOATs, and compared the activities of MRP2 toward MPAG in rats and humans to clarify the cause of the species difference in excretion of MPAG.
Materials and METHODS
Materials
MPAG was kindly supplied by Roche Palo Alto (CA, USA). MPA was purchased from Wako Pure Chemicals (Osaka, Japan). p-[glycyl-2-3H]-Aminohippuric acid (PAH) (156 GBq/mmol) and [6,7-3H(N)]-Estron sulfate, ammonium salt (ES) (2,120 GBq/mmol) was purchased from Perkin Elmer Life Sciences (Boston, MA). Cephaloridine was kindly supplied by Shionogi & Co., Ltd (Osaka, Japan). Human MRP2 (hMRP2) vesicles and Rat Mrp2 (rMrp2) vesicles, inside-out vesicles of Sf9 cells expressing hMRP2 and rMrp2, respectively, were purchased from GenoMembrane (Osaka, Japan). All other reagents were of the highest grade available.
Functional expression of hOAT1 in HeLa cells
Functional expression of hOAT1 in HeLa cells was done using the procedure described by Ganapathy et al. with some modifications [17]. Subconfluent HeLa cells in 24-well culture plates were first infected with a recombinant vaccinia virus, VTF7–3 (American Type Culture Collection, Manassas, VA), that carries the gene for T7 RNA polymerase as a part of its genome. This enables the HeLa cells to express T7 RNA polymerase. Following the infection, the cells were transfected using lipofectin® (Invitrogen, Carlsbad, CA, USA) with pcDNA3.1(+) (Invitrogen)-hOAT1 (SLC22A6, transcript variant 2, Genebank accession number: NM_153276) construct that had been subcloned previously [18]. In this construct, the cDNA was under control of T7 promoter in the plasmid. Cells transfected with an empty plasmid served as control cells. Transfection was mediated by lipofection. The virus-encoded T7 RNA polymerase catalyzes transcription of the cDNA, allowing transient expression of the hOAT1 protein in the HeLa cell plasma membrane. At 12 h after infection, uptake measurements were made at room temperature. Uptake experiments were carried out as described previously with minor modification [18]. The uptake buffer consisted of 137 mM NaCl, 5.36 mM KCl, 1.26 mM CaCl2, 0.81 mM MgSO4, 0.44 mM KH2PO4, 0.34 mM Na2HPO4, 25 mM D-glucose, and 10 mM HEPES/Tris (pH 7.4). After washing the cells with 1 mL of the uptake buffer, uptake was started by adding 0.25 mL of substrate solution containing 0.25 µCi [3H]-PAH (240 nM). The time of incubation for uptake measurements was 15 min. At the end of the incubation, the uptake was terminated by aspiration of the substrate solution followed by washing twice with 1 mL of ice-cold transport buffer. The cells were lysed with 0.25 mL of 1% SDS in 0.2 M NaOH, and the radioactivity was measured. A small portion of the cell lysate was used for the determination of protein concentration. Uptake values are expressed as pmol/mg protein. hOAT1-specific transport was calculated by subtracting the transport in vector-transfected cells from the transport in cDNA-transfected cells.
Expression and transport assay in Xenopus laevis oocytes
A full-length clone of hOAT3 was obtained by RT-PCR from human kidney total RNA. After reverse transcription (42ºC, 60 min) using oligo (dT) primer and ReverTra Ace (TOYOBO, Osaka, Japan), amplification was performed for 35 cycles of 94ºC for 30 sec, 54ºC for 30 sec, and 72ºC for 90 sec using Pyrobest DNA polymerase (Takara, Osaka, Japan). Forward and reverse primers were 5’-tgccatgaccttctcggaga-3’ and 5’-ttccgttgtcctcagctgga-3’, respectively. The obtained fragments with blunt ends were purified and subcloned into pCR-Blunt II-TOPO (Invitrogen, Carlsbad. CA). The sequence was analyzed using an ABI310 sequencer (Applied Biosystems, Foster City, CA). The coding sequence was identical to the published sequence of hOAT3 (SLC22A8, Genebank accession number: NM_004254). The hOAT3 cDNA fragment was obtained by digestion of a pCR-Blunt II-TOPO-hOAT cDNA construct with Hind III and EcoR V (Takara, Osaka, Japan). The fragment was subcloned into pcDNA3.1(+). This construct was used for the expression of hOAT3 in HeLa cells. The human OAT3 clone was also amplified using a forward primer including EcoR I digestion site (5’-ggattcccaccatgaccttctcgga-3’) and reverse primer including the Xba I digestion site (5’-tctagatcagctggagcccagg-3’). Amplification was performed for 30 cycles of 98ºC for 10 sec, 55ºC for 30 sec, and 72ºC for 120 sec using Pyrobest DNA polymerase. This PCR product was digested by Eco RI and Xba I, and then the fragment was subcloned into pGH19 (generous gift from Dr. Peter S. Aronson, Department of Internal Medicine, Yale University, School of Medicine, New Haven, CT), which is also digested by the same restriction enzymes. These constructs were used for synthesis of cRNA for expression in Xenopus oocytes. hOAT1- pcDNA3.1(+), mentioned above, was digested by Xba I and Eco RI, and then the fragment was subcloned into pGH19. cRNA was synthesized from linearized plasmids with T7 RNA polymerase, and a poly(A) tail was added by using an mMESSAGE mMACHINE and Poly(A) Tailing Kit (Ambion, Austin, TX). Mature oocytes from Xenopus laevis were isolated by treatment with 2 mg/mL of collagenase (Wako, Okasa, Japan), manually defolliculated, and maintained at 16℃ in modified Barth’s medium supplemented with 50 mg/L of gentamicin. On the following day, oocytes were microinjected with either 50 nL of water containing 50 ng cRNA or 50 nL water alone. Uptake of ES was measured 3 days after microinjection. Uptake experiments were performed at room temperature. The transport buffer used in this study consisted of 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2 and 5 mM HEPES/Tris (pH 7.4). Oocytes were incubated in 100 μl of transport buffer containing 0.35 μCi [3H]-ES (17.5 nM) for hOAT3 uptake assay. At the end of incubation, oocytes were washed five times with ice-cold transport buffer. Then oocytes were dissolved in 10% SDS solution and radioactivity was measured by a liquid scintillation counter.
Two-microelectrode voltage-clamp technique
Electrophysiological studies were done by the conventional two-microelectrode voltage-clamp method according to a previous report [19]. Oocytes expressing hOAT1 or hOAT3 were perfused with various concentrations of tested drugs at pH 7.4, and the inward currents induced by the substrate flux were monitored under voltage-clamp conditions (-50 mV). For studies involving the current-membrane voltage relationship, step changes in membrane potential were applied each for a duration of 100 ms in 20 mV increments. The buffer used in this study was the same as the transport buffer used for transport assay. The magnitude of tested drug-induced current was considered as a measure of transport rate.
Uptake experiments using membrane vesicles expressing hMRP2 or rMrp2
Ten µL (50 µg) of membrane vesicle suspension was preincubated for 1 minute at 37℃. The uptake was initiated by adding 250 µL of reaction solution consisting of 1 µCi (960 nM) [3H]-PAH, 70 mM KCl, 7.5 mM MgCl2, 4 mM MgATP or MgAMP, and 50 mM MOPS/Tris (pH 7.0) with MPAG of various concentrations. After 30 minutes, the reaction was terminated by diluting the reaction mixture with 3 mL of an ice-cold stop buffer (70 mM KCl, 40 mM MOPS/Tris, pH 7.0) followed by filtration through a Millipore filter (HAWP, 0.45 µm. 2.5 cm in diameter). The filter was then washed twice with 8 mL of the same ice-cold stop buffer. Radioactivity was measured by a liquid scintillation counter.
Animal Experiments
Male Wistar rats were obtained from Hokudo (Sapporo, Japan). Male Eisai hyperbilirubinuria rats (EHBRs) were purchased from Sankyo Labo Service (Tokyo, Japan). The body weights of rats used in this study were 240 to 350 g. The experimental protocols were reviewed and approved by the Hokkaido University Animal Care Committee in accordance with the “Guide for the Care and Use of Laboratory Animals”.
Animal Experiments were done according to the report of Kobayashi et al. [13]. Rats were anesthetized with an intraperitoneal injection of sodium pentobarbital (40 mg/kg). After abdominal operation, a polyethylene tube was inserted into the bile duct toward the liver and then MPA, which was dissolved in polyethylene glycol 400 at a concentration of 5 mg/mL, was administered intravenously to each rat via the femoral vein. The dose of MPA was fixed at 5 mg/kg body weight. Bile samples were collected at 10, 20 and 30 minutes after administration, and the bile volume was measured with an appropriately sized volumetric pipette. Bile samples were kept frozen until assay.
Determination of MPAG concentration in bile
Plasma concentrations of MPAG were determined by reversed-phase high-performance liquid chromatography (HPLC) with an ultraviolet detector. The separation was performed on an ERC ODS-1161 (6.0 mm I.D. x 100 mm) column (YOKOHAMARIKA Co., Yokohama, Japan). The mobile phase was a mixture of acetonitrile and 60 mM phosphoric acid (23:77). The flow rate was 1.0 mL/min and column temperature was 55ºC. Wavelengths of 265 nm were used for ultraviolet detection.
Twenty µL of bile was mixed with 20 µL of H2O, 40 µL of 2% H3PO4 and 10 µL of ß-naphthol solution (50 µg/mL in methanol) as an internal standard and then vortexed for 20 sec. Forty µL of the mixture was injected into the HPLC system. The lower limit of quantification for MPAG was 1.5 µM. Coefficients of variation were 2.17% and 1.31% at 500 µM and 1.5 µM, respectively (n = 5).
Statistical Analysis
Data are expressed as mean±S.E. IC50 values were calculated with logistic function using Origin version 6.1 (OriginLab, Northampton, MA).
RESULTS
Inhibitory effect of MPAG on the uptake of PAH by HeLa-hOAT1 cells
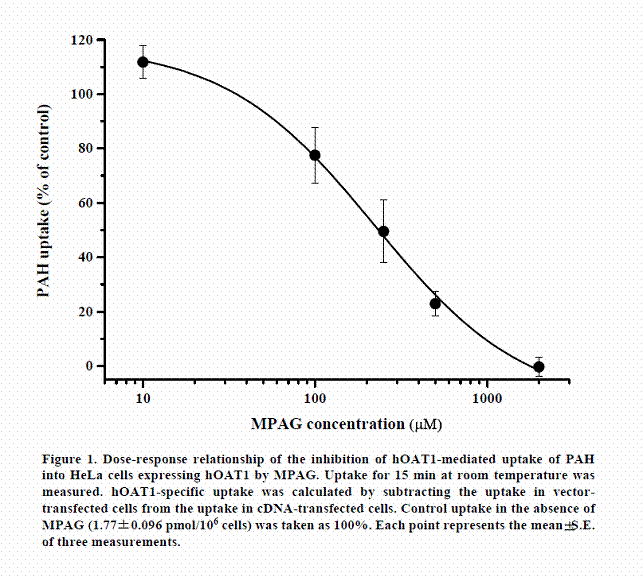
We examined whether active transports participated in renal excretion of MPAG in humans. First, the effect of MPAG on the uptake of PAH (a typical substrate for hOAT1) by HeLa-hOAT1 cells was examined. The dose-response relationship for the inhibition of PAH uptake by MPAG is shown in Figure 1. MPAG inhibited the uptake of PAH, and the calculated IC50 value was 222.6± 26.6 µM. This result suggests that MPAG is able to be a inhibitor or substrate of hOAT1.
Inhibitory effect of MPAG on the uptake of ES by Xenopus laevis oocytes expressing hOAT3
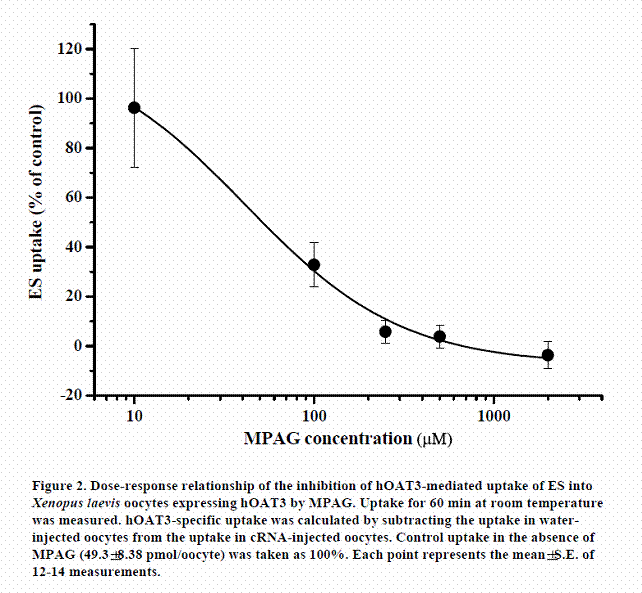
In the same way, we examined whether MPAG inhibited uptake of the substrate via hOAT3. The inhibitory effect of MPAG on the uptake of ES (a typical substrate for hOAT3) by Xenopus laevis oocytes expressing hOAT3 was examined. The dose-response relationship for the inhibition of ES uptake by MPAG is shown in Figure 2. MPAG remarkably inhibited the uptake of ES, and the calculated IC50 value was 41.5±11.5 µM. The inhibitory effect of MPAG for hOAT3 was stronger than that for hOAT1. This result suggests that MPAG is able to be an inhibitor or substrate of hOAT3 and that affinity of MPAG for hOAT3 is higher than that for hOAT1.
Measurement of transport-induced current using the two-microelectrode voltage-clamp technique in Xenopus laevis oocytes expressing hOATs
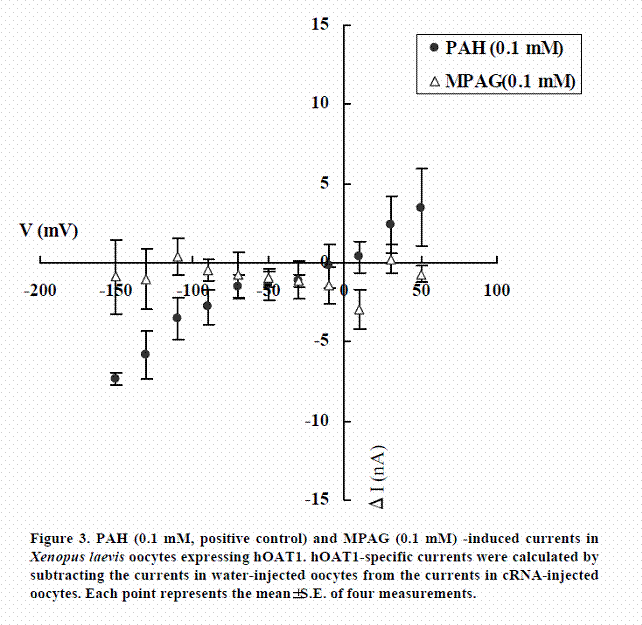
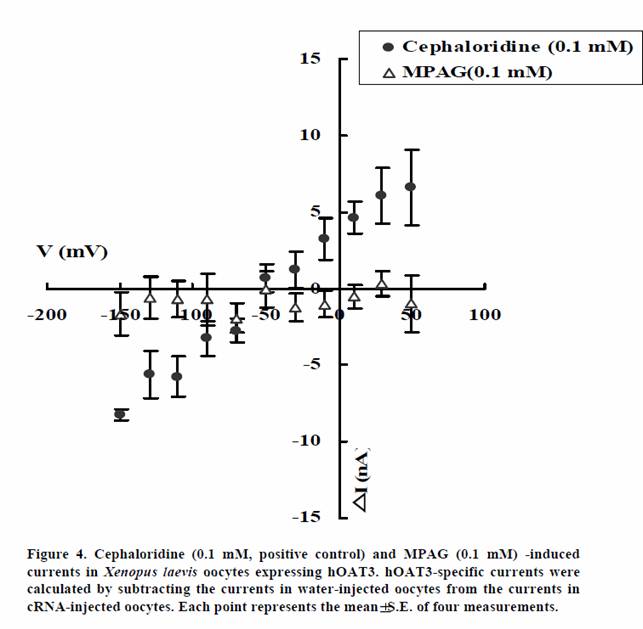
We examined whether MPAG is not only inhibitor of hOAT1 and hOAT3 but also substrate. The transport activities of MPAG via hOAT1 and hOAT3 were studied by electrophysiological methods. The activities were assessed by monitoring currents induced by the tested drugs in hOAT1- or hOAT3-expressing oocytes under voltage-clamp conditions. PAH and cephaloridine were used in this study as positive controls for hOAT1 and hOAT3, respectively. As shown in Figures 3 and 4, PAH and cephaloridine induced currents in hOAT1- and hOAT3-expressing oocytes, respectively. On the other hand, MPAG did not induce any current. These results suggest that MPAG is not excreted into urine via hOAT1 and hOAT3 in human or MPAG is transported via hOAT1 and hOAT3 with electroneutral manner.
Inhibitory effect of MPAG on the uptake of PAH via hMRP2 and rMrp2
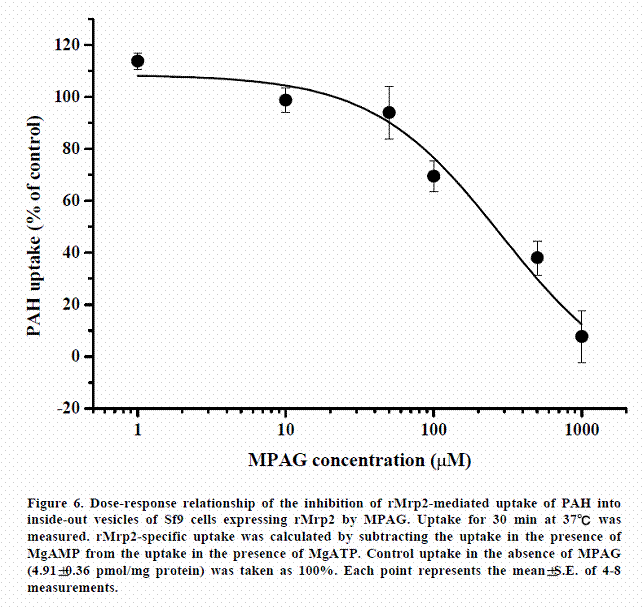
In order to compare the affinity of MPAG transport activity of hMRP2 with that of rMrp2, we examined the inhibitory effect of MPAG on the uptake of PAH (a typical substrate for MRP2) via hMRP2 and that via rMrp2. As shown in Figures 5 and 6, MPAG strongly inhibited the uptake of PAH both via rMrp2 and hMRP2. However, the magnitudes of the inhibitory effects were different. The calculated IC50 values in rMrp2 and hMRP2 were 286.2±157.3 µM and 1036.8±330.5 µM, respectively. Namely, affinity of MPAG for MRP2 is higher in rats than in humans.
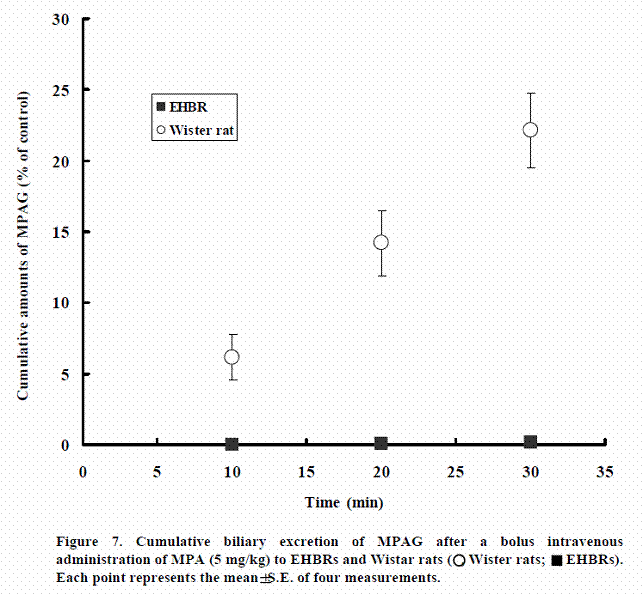
Biliary excretion of MPAG after intravenous administration of MPA to Wister rat and EHBR
Figure 7 shows the cumulative excretion of MPAG after bolus intravenous administration of MPA to Wistar rats and EHBRs genetically lacking rMrp2. In Wistar rats, about 22.1% of the dose of MPA had been excreted as MPAG into bile at 30 min after administration. In contrast, MPAG was not excreted into bile in EHBRs.
These results suggest that only Mrp2 is responsible for excretion of MPAG into bile.
DISCUSSION
Generally, rats are used to study pharmacokinetics of drugs. However, pharmacokinetics in rats often do not correspond with that in humans. MPA is one such drug [20]. In humans, MPAG, which is the main metabolite of MPA, is mostly excreted into urine [10], whereas more than 80% of the dose is excreted into bile in rats [12]. MPAG is produced by direct glucuronidation of MPA and is excreted into bile. It is hydrolyzed to MPA by ß-glucuronidase in the intestine, and the MPA produced is then absorbed again into the blood stream (enterohepatic circulation) [6]. The amounts of re-absorbed MPA affect pharmacokinetics of MPA, and especially, AUC depends on enterohepatic circulation [21]. Therefore, it is important to clarify the cause of the difference between MPAG elimination in humans and rats. We planned this study to clarify the causes of this species difference with two hypotheses: 1) in humans, MPAG may be excreted by active transport in the kidney and 2) the affinity of rMrp2 for MPAG may be higher than that of hMRP2.
In proximal tubules, many organic anions are secreted in two transmembrane transport steps. First, organic anions are taken up from peritubular plasma by basolateral organic anion transporters, and then organic anions are exported into the tubular lumen by other transporters expressed in the epithelial brush-border membrane [15, 22]. OATs transport not only many kinds of organic anionic drugs (such as antibiotics, NSAIDs, and ACE inhibitors) but also some conjugated substances (such as sulphate and glucuronide). OAT1 and OAT3 have been reported to be mainly responsible for renal excretion of organic anionic compounds [14-16]. Therefore, we investigated whether MPAG is excreted into urine via hOAT1 and hOAT3.
In this study, MPAG inhibited both PAH uptake via hOAT1 and ES uptake via hOAT3. The calculated IC50 values of MPAG forvhOAT1 and hOAT3 were 222.6± 26.6 µM and 41.5±11.5 µM, respectively. The maximum concentration of MPAG in patients administered MMF after renal transplantation in our hospital was about 50 to 200 µM (data not shown). Since most patients take ACE inhibitors, antibiotics, and/or anti-viral drugs together with immunosuppresants, it is possible that drug-drug interaction occurs in renal excretion via OAT1 and OAT3 in humans.
We then investigated whether MPAG is also a substrate for hOAT1 and hOAT3. It has been reported that OAT1 and OAT3 are sodium-independent anion/dicarboxylate exchangers that have electrogenic properties [23, 24]. Therefore, we investigated whether MPAG is actually transported via hOAT1 and hOAT3 by the two-microelectrode voltage-clamp technique using Xenopus laevis oocytes expressing hOATs. Neither hOAT1-expressing nor hOAT3-expressing oocytes showed MPAG-induced currents. These results demonstrated that MPAG is not a substrate but is an inhibitor for hOAT1 and hOAT3 or MPAG is transported via hOAT1 and hOAT3 with electroneutral manner.
It is known that MPAG is transported from hepatocytes to the canalicular lumen by MRP2. We therefore investigated the transport of MPAG by MRP2 to determine whether there are any differences in affinity of MRP2 in MPAG transport between rats and humans. In this study, the affinity of rMRP2 to MPAG was about 3.6-times higher than that of hMRP2 (Figures 5 and 6). In addition, excretion of MPAG into bile was hardly observed in EHBRs (which genetically lack Mrp2), as already reported [13]. These results suggest that MPAG could not be excreted into bile without Mrp2 and that the difference in affinity between hMRP2 and rMrp2 is a cause of the difference in the excretion ratio of MPAG. Kuroda et al. reported that hepatic expression of Mrp3 was increased in EHBRs [25]. Mrp3 is capable of transporting several glucuronide conjugates from hepatocytes into sinusoidal blood [26]. Kobayashi et al. reported that MPAG did not appear in plasma of Sprague-Dawley (SD) rats after intravenous administration of MPAG in contrast to EHBRs and Wistar rats and that Mrp3 probably contributed to the appearance of MPAG in plasma [13]. However, the excretion ratio of MPAG was lower in SD rats than in Wistar rats, though MPAG did not appear in the blood stream in their study. Furthermore, differences in the affinities of 17ß-estradiol 17-(ß-D-glucuronide) and leukotriene C4 for Mrp2 were observed between rats and dogs [27]. These results indicate that differences in the excretion ratio of MPAG into bile between species are caused by the difference in affinities of MPAG for Mrp2 between species. This study demonstrated that difference between pharmacokinetics of MPA in rats and that in humans is caused by different affinities of MRP2 to a glucuronized form. Therefore, the result suggested that pharmacokinetics of MPA are able to change in patients who have hMRP2 with low affinity caused by genetic polymorphisms.
CONCLUSIONS
The aim of this study was to clarify the cause of interspecies variability (humans and rats) of MPA pharmacokinetics. MPAG is not a substrate but is an inhibitor for hOAT1 and hOAT3. The affinity of rMrp2 to MPAG is about 3.6-times higher than that of hMRP2. Therefore, the difference in affinity between hMRP2 and rMrp2 is a cause of the difference in excretion ratio of MPAG.